Yingnan Liu, Dashuai Wang, Bin Yang, Zhongjian Li, Tao Zhang, Raul D. Rodriguez, Lecheng Lei and Yang Hou
{"title":"高通量筛选串联催化剂将二氧化碳还原为多碳产品的机理见解","authors":"Yingnan Liu, Dashuai Wang, Bin Yang, Zhongjian Li, Tao Zhang, Raul D. Rodriguez, Lecheng Lei and Yang Hou","doi":"10.1039/D4CP01622J","DOIUrl":null,"url":null,"abstract":"<p >In carbon dioxide electrochemical reduction (CO<small><sub>2</sub></small>ER), since isolated catalysts encounter challenges in meeting the demands of intricate processes for producing multi-carbon (C<small><sub>2+</sub></small>) products, tandem catalysis is emerging as a promising approach. Nevertheless, there remains an insufficient theoretical understanding of designing tandem catalysts. Herein, we utilized density functional theory (DFT) to screen 80 tandem catalysts for efficient CO<small><sub>2</sub></small>ER to C<small><sub>2</sub></small> products systematically, which combines the advantages of nitrogen-doped carbon-supported transition metal single-atom catalysts (M–N–C) and copper clusters. Three crucial criteria were designed to select structures for generation and transfer of *CO and facilitate C–C coupling. The optimal Cu/RuN<small><sub>4</sub></small>-pl catalyst exhibited an excellent ethanol production capacity. Additionally, the relationship between CO adsorption strength and transfer energy barrier was established, and the influence of the electronic structure on its adsorption strength was studied. This provided a novel and well-considered solution and theoretical guidance for the design of rational composition and structurally superior tandem catalysts.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 30","pages":" 20399-20408"},"PeriodicalIF":2.9000,"publicationDate":"2024-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mechanistic insights into high-throughput screening of tandem catalysts for CO2 reduction to multi-carbon products†\",\"authors\":\"Yingnan Liu, Dashuai Wang, Bin Yang, Zhongjian Li, Tao Zhang, Raul D. Rodriguez, Lecheng Lei and Yang Hou\",\"doi\":\"10.1039/D4CP01622J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In carbon dioxide electrochemical reduction (CO<small><sub>2</sub></small>ER), since isolated catalysts encounter challenges in meeting the demands of intricate processes for producing multi-carbon (C<small><sub>2+</sub></small>) products, tandem catalysis is emerging as a promising approach. Nevertheless, there remains an insufficient theoretical understanding of designing tandem catalysts. Herein, we utilized density functional theory (DFT) to screen 80 tandem catalysts for efficient CO<small><sub>2</sub></small>ER to C<small><sub>2</sub></small> products systematically, which combines the advantages of nitrogen-doped carbon-supported transition metal single-atom catalysts (M–N–C) and copper clusters. Three crucial criteria were designed to select structures for generation and transfer of *CO and facilitate C–C coupling. The optimal Cu/RuN<small><sub>4</sub></small>-pl catalyst exhibited an excellent ethanol production capacity. Additionally, the relationship between CO adsorption strength and transfer energy barrier was established, and the influence of the electronic structure on its adsorption strength was studied. This provided a novel and well-considered solution and theoretical guidance for the design of rational composition and structurally superior tandem catalysts.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 30\",\"pages\":\" 20399-20408\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-07-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01622j\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01622j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
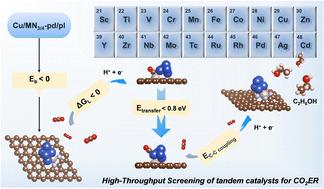
Mechanistic insights into high-throughput screening of tandem catalysts for CO2 reduction to multi-carbon products†
In carbon dioxide electrochemical reduction (CO2ER), since isolated catalysts encounter challenges in meeting the demands of intricate processes for producing multi-carbon (C2+) products, tandem catalysis is emerging as a promising approach. Nevertheless, there remains an insufficient theoretical understanding of designing tandem catalysts. Herein, we utilized density functional theory (DFT) to screen 80 tandem catalysts for efficient CO2ER to C2 products systematically, which combines the advantages of nitrogen-doped carbon-supported transition metal single-atom catalysts (M–N–C) and copper clusters. Three crucial criteria were designed to select structures for generation and transfer of *CO and facilitate C–C coupling. The optimal Cu/RuN4-pl catalyst exhibited an excellent ethanol production capacity. Additionally, the relationship between CO adsorption strength and transfer energy barrier was established, and the influence of the electronic structure on its adsorption strength was studied. This provided a novel and well-considered solution and theoretical guidance for the design of rational composition and structurally superior tandem catalysts.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
