{"title":"设计金属有机框架的有机桥接连接体以增强二氧化碳吸附能力","authors":"Kahkasha Parveen and Srimanta Pakhira","doi":"10.1039/D4NJ01197J","DOIUrl":null,"url":null,"abstract":"<p >The global rate of anthropogenic carbon dioxide (CO<small><sub>2</sub></small>) emission is rising, which urges the development of efficient carbon capture and storage (CCS) technologies. Among the various CO<small><sub>2</sub></small> capture methods, adsorption by metal–organic framework (MOF) linkers as excellent CO<small><sub>2</sub></small> adsorbents has attracted immense interest because of their important role in understanding the interaction mechanism for CO<small><sub>2</sub></small> adsorption. Here, we have investigated the adsorption of a CO<small><sub>2</sub></small> molecule at the center and side positions of eight MOF-linkers using molecular cluster models. The interaction between CO<small><sub>2</sub></small> and the linkers is assessed by computing the binding enthalpy (Δ<em>H</em>) through the first principles-based density functional theory (DFT) with Grimme's dispersion corrections (<em>i.e.</em>, B3LYP-D3) and second-order Møller Plesset theory (MP2) methods. The results of our investigations revealed that the center and side positions of the FBDC, DFBDC-1, DFBDC-2, and TFBDC linkers, the side position of the DClBDC-2 linker, and the center position of the NDC linkers exhibit favorable physisorption behavior for CO<small><sub>2</sub></small> adsorption, with the values of Δ<em>H</em> ranging from −12.05 to −14.09 kJ mol<small><sup>−1</sup></small>. After lithium (Li) decoration on the pure linkers, CO<small><sub>2</sub></small> adsorption at the center position of the BDC, FBDC, and DClBDC-1 linkers and the side position of the BDC, FBDC, DFBDC-1, and DFBDC-2 linkers reflects a strong physisorption behavior with the values of Δ<em>H</em> lying in the range of −34.64 to −35.30 kJ mol<small><sup>−1</sup></small> but remaining below the energy of a chemical bond (chemisorption), which is required for facile CO<small><sub>2</sub></small> release. To support our computed results, energy decomposition analysis (EDA) has been performed and the EDA study reveals that among all the energy components, the contribution of electrostatic and polarization energy components to the Δ<em>H</em> value is the most dominant. Frontier molecular orbital (FMO) analysis demonstrated the stability of the Li-decorated linkers. The results of our investigations will direct the development and synthesis of novel porous MOFs with enhanced CO<small><sub>2</sub></small> adsorption.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 31","pages":" 13700-13714"},"PeriodicalIF":2.5000,"publicationDate":"2024-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Designing organic bridging linkers of metal–organic frameworks for enhanced carbon dioxide adsorption†\",\"authors\":\"Kahkasha Parveen and Srimanta Pakhira\",\"doi\":\"10.1039/D4NJ01197J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The global rate of anthropogenic carbon dioxide (CO<small><sub>2</sub></small>) emission is rising, which urges the development of efficient carbon capture and storage (CCS) technologies. Among the various CO<small><sub>2</sub></small> capture methods, adsorption by metal–organic framework (MOF) linkers as excellent CO<small><sub>2</sub></small> adsorbents has attracted immense interest because of their important role in understanding the interaction mechanism for CO<small><sub>2</sub></small> adsorption. Here, we have investigated the adsorption of a CO<small><sub>2</sub></small> molecule at the center and side positions of eight MOF-linkers using molecular cluster models. The interaction between CO<small><sub>2</sub></small> and the linkers is assessed by computing the binding enthalpy (Δ<em>H</em>) through the first principles-based density functional theory (DFT) with Grimme's dispersion corrections (<em>i.e.</em>, B3LYP-D3) and second-order Møller Plesset theory (MP2) methods. The results of our investigations revealed that the center and side positions of the FBDC, DFBDC-1, DFBDC-2, and TFBDC linkers, the side position of the DClBDC-2 linker, and the center position of the NDC linkers exhibit favorable physisorption behavior for CO<small><sub>2</sub></small> adsorption, with the values of Δ<em>H</em> ranging from −12.05 to −14.09 kJ mol<small><sup>−1</sup></small>. After lithium (Li) decoration on the pure linkers, CO<small><sub>2</sub></small> adsorption at the center position of the BDC, FBDC, and DClBDC-1 linkers and the side position of the BDC, FBDC, DFBDC-1, and DFBDC-2 linkers reflects a strong physisorption behavior with the values of Δ<em>H</em> lying in the range of −34.64 to −35.30 kJ mol<small><sup>−1</sup></small> but remaining below the energy of a chemical bond (chemisorption), which is required for facile CO<small><sub>2</sub></small> release. To support our computed results, energy decomposition analysis (EDA) has been performed and the EDA study reveals that among all the energy components, the contribution of electrostatic and polarization energy components to the Δ<em>H</em> value is the most dominant. Frontier molecular orbital (FMO) analysis demonstrated the stability of the Li-decorated linkers. The results of our investigations will direct the development and synthesis of novel porous MOFs with enhanced CO<small><sub>2</sub></small> adsorption.</p>\",\"PeriodicalId\":95,\"journal\":{\"name\":\"New Journal of Chemistry\",\"volume\":\" 31\",\"pages\":\" 13700-13714\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"New Journal of Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/nj/d4nj01197j\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/nj/d4nj01197j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Designing organic bridging linkers of metal–organic frameworks for enhanced carbon dioxide adsorption†
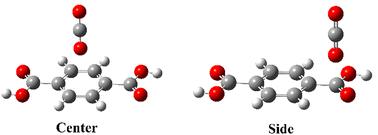
The global rate of anthropogenic carbon dioxide (CO2) emission is rising, which urges the development of efficient carbon capture and storage (CCS) technologies. Among the various CO2 capture methods, adsorption by metal–organic framework (MOF) linkers as excellent CO2 adsorbents has attracted immense interest because of their important role in understanding the interaction mechanism for CO2 adsorption. Here, we have investigated the adsorption of a CO2 molecule at the center and side positions of eight MOF-linkers using molecular cluster models. The interaction between CO2 and the linkers is assessed by computing the binding enthalpy (ΔH) through the first principles-based density functional theory (DFT) with Grimme's dispersion corrections (i.e., B3LYP-D3) and second-order Møller Plesset theory (MP2) methods. The results of our investigations revealed that the center and side positions of the FBDC, DFBDC-1, DFBDC-2, and TFBDC linkers, the side position of the DClBDC-2 linker, and the center position of the NDC linkers exhibit favorable physisorption behavior for CO2 adsorption, with the values of ΔH ranging from −12.05 to −14.09 kJ mol−1. After lithium (Li) decoration on the pure linkers, CO2 adsorption at the center position of the BDC, FBDC, and DClBDC-1 linkers and the side position of the BDC, FBDC, DFBDC-1, and DFBDC-2 linkers reflects a strong physisorption behavior with the values of ΔH lying in the range of −34.64 to −35.30 kJ mol−1 but remaining below the energy of a chemical bond (chemisorption), which is required for facile CO2 release. To support our computed results, energy decomposition analysis (EDA) has been performed and the EDA study reveals that among all the energy components, the contribution of electrostatic and polarization energy components to the ΔH value is the most dominant. Frontier molecular orbital (FMO) analysis demonstrated the stability of the Li-decorated linkers. The results of our investigations will direct the development and synthesis of novel porous MOFs with enhanced CO2 adsorption.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
