Shasha Tian , Xiaopeng Yang , Yao Lin , Xinran Li , Saijun Zhou , Pei Yu , Yanjun Zhao
{"title":"PDK4 介导的 Nrf2 失活导致了氧化应激和糖尿病肾损伤。","authors":"Shasha Tian , Xiaopeng Yang , Yao Lin , Xinran Li , Saijun Zhou , Pei Yu , Yanjun Zhao","doi":"10.1016/j.cellsig.2024.111282","DOIUrl":null,"url":null,"abstract":"<div><p>Diabetic kidney disease (DKD) is often featured with redox dyshomeostatis. Pyruvate dehydrogenase kinase 4 (PDK4) is the hub for DKD development. However, the mechanism by which PDK4 mediates DKD is poorly understood. The current work aimed to elucidate the relationship between PDK4 and DKD from the perspective of redox manipulation. Oxidative stress was observed in the human proximal tubular cell line (HK-2 cells) treated with a high concentration of glucose and palmitic acid (HGL). The mechanistic study showed that PDK4 could upregulate Kelch-like ECH-associated protein 1 (Keap1) in HGL-treated HK-2 cells through the suppression of autophagy, resulting in the depletion of nuclear factor erythroid 2-related factor 2 (Nrf2), the master regulator of redox homeostasis. At the cellular level, pharmacological inhibition or genetic knockdown of PDK4 could boost Nrf2, followed by the increase of a plethora of antioxidant enzymes and ferroptosis-suppression enzymes. Meanwhile, the inhibition or knockdown of PDK4 remodeled iron metabolism, further mitigating oxidative stress and lipid peroxidation. The same trend was observed in the DKD mice model. The current work highlighted the role of PDK4 in the development of DKD and suggested that PDK4 might be a promising target for the management of DKD.</p></div>","PeriodicalId":9902,"journal":{"name":"Cellular signalling","volume":"121 ","pages":"Article 111282"},"PeriodicalIF":3.7000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"PDK4-mediated Nrf2 inactivation contributes to oxidative stress and diabetic kidney injury\",\"authors\":\"Shasha Tian , Xiaopeng Yang , Yao Lin , Xinran Li , Saijun Zhou , Pei Yu , Yanjun Zhao\",\"doi\":\"10.1016/j.cellsig.2024.111282\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Diabetic kidney disease (DKD) is often featured with redox dyshomeostatis. Pyruvate dehydrogenase kinase 4 (PDK4) is the hub for DKD development. However, the mechanism by which PDK4 mediates DKD is poorly understood. The current work aimed to elucidate the relationship between PDK4 and DKD from the perspective of redox manipulation. Oxidative stress was observed in the human proximal tubular cell line (HK-2 cells) treated with a high concentration of glucose and palmitic acid (HGL). The mechanistic study showed that PDK4 could upregulate Kelch-like ECH-associated protein 1 (Keap1) in HGL-treated HK-2 cells through the suppression of autophagy, resulting in the depletion of nuclear factor erythroid 2-related factor 2 (Nrf2), the master regulator of redox homeostasis. At the cellular level, pharmacological inhibition or genetic knockdown of PDK4 could boost Nrf2, followed by the increase of a plethora of antioxidant enzymes and ferroptosis-suppression enzymes. Meanwhile, the inhibition or knockdown of PDK4 remodeled iron metabolism, further mitigating oxidative stress and lipid peroxidation. The same trend was observed in the DKD mice model. The current work highlighted the role of PDK4 in the development of DKD and suggested that PDK4 might be a promising target for the management of DKD.</p></div>\",\"PeriodicalId\":9902,\"journal\":{\"name\":\"Cellular signalling\",\"volume\":\"121 \",\"pages\":\"Article 111282\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cellular signalling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S089865682400250X\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular signalling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S089865682400250X","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/5 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
PDK4-mediated Nrf2 inactivation contributes to oxidative stress and diabetic kidney injury
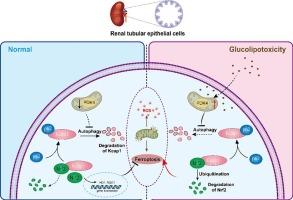
Diabetic kidney disease (DKD) is often featured with redox dyshomeostatis. Pyruvate dehydrogenase kinase 4 (PDK4) is the hub for DKD development. However, the mechanism by which PDK4 mediates DKD is poorly understood. The current work aimed to elucidate the relationship between PDK4 and DKD from the perspective of redox manipulation. Oxidative stress was observed in the human proximal tubular cell line (HK-2 cells) treated with a high concentration of glucose and palmitic acid (HGL). The mechanistic study showed that PDK4 could upregulate Kelch-like ECH-associated protein 1 (Keap1) in HGL-treated HK-2 cells through the suppression of autophagy, resulting in the depletion of nuclear factor erythroid 2-related factor 2 (Nrf2), the master regulator of redox homeostasis. At the cellular level, pharmacological inhibition or genetic knockdown of PDK4 could boost Nrf2, followed by the increase of a plethora of antioxidant enzymes and ferroptosis-suppression enzymes. Meanwhile, the inhibition or knockdown of PDK4 remodeled iron metabolism, further mitigating oxidative stress and lipid peroxidation. The same trend was observed in the DKD mice model. The current work highlighted the role of PDK4 in the development of DKD and suggested that PDK4 might be a promising target for the management of DKD.
期刊介绍:
Cellular Signalling publishes original research describing fundamental and clinical findings on the mechanisms, actions and structural components of cellular signalling systems in vitro and in vivo.
Cellular Signalling aims at full length research papers defining signalling systems ranging from microorganisms to cells, tissues and higher organisms.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
