Wenhao Jing, Zihao Jiao, Mengmeng Song, Ya Liu, Liejin Guo
{"title":"基于局部电子转移特征预测二元氧化物上氢原子吸附能的主动学习工作流程","authors":"Wenhao Jing, Zihao Jiao, Mengmeng Song, Ya Liu, Liejin Guo","doi":"10.1016/j.gee.2024.06.007","DOIUrl":null,"url":null,"abstract":"Machine learning combined with density functional theory (DFT) enables rapid exploration of catalyst descriptors space such as adsorption energy, facilitating rapid and effective catalyst screening. However, there is still a lack of models for predicting adsorption energies on oxides, due to the complexity of elemental species and the ambiguous coordination environment. This work proposes an active learning workflow (LeNN) founded on local electronic transfer features () and the principle of coordinate rotation invariance. By accurately characterizing the electron transfer to adsorption site atoms and their surrounding geometric structures, LeNN mitigates abrupt feature changes due to different element types and clarifies coordination environments. As a result, it enables the prediction of ∗H adsorption energy on binary oxide surfaces with a mean absolute error (MAE) below 0.18 eV. Moreover, we incorporate local coverage () and leverage neutral network ensemble to establish an active learning workflow, attaining a prediction MAE below 0.2 eV for 5419 multi-∗H adsorption structures. These findings validate the universality and capability of the proposed features in predicting ∗H adsorption energy on binary oxide surfaces.","PeriodicalId":12744,"journal":{"name":"Green Energy & Environment","volume":"40 1","pages":""},"PeriodicalIF":14.6000,"publicationDate":"2024-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"An active learning workflow for predicting hydrogen atom adsorption energies on binary oxides based on local electronic transfer features\",\"authors\":\"Wenhao Jing, Zihao Jiao, Mengmeng Song, Ya Liu, Liejin Guo\",\"doi\":\"10.1016/j.gee.2024.06.007\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Machine learning combined with density functional theory (DFT) enables rapid exploration of catalyst descriptors space such as adsorption energy, facilitating rapid and effective catalyst screening. However, there is still a lack of models for predicting adsorption energies on oxides, due to the complexity of elemental species and the ambiguous coordination environment. This work proposes an active learning workflow (LeNN) founded on local electronic transfer features () and the principle of coordinate rotation invariance. By accurately characterizing the electron transfer to adsorption site atoms and their surrounding geometric structures, LeNN mitigates abrupt feature changes due to different element types and clarifies coordination environments. As a result, it enables the prediction of ∗H adsorption energy on binary oxide surfaces with a mean absolute error (MAE) below 0.18 eV. Moreover, we incorporate local coverage () and leverage neutral network ensemble to establish an active learning workflow, attaining a prediction MAE below 0.2 eV for 5419 multi-∗H adsorption structures. These findings validate the universality and capability of the proposed features in predicting ∗H adsorption energy on binary oxide surfaces.\",\"PeriodicalId\":12744,\"journal\":{\"name\":\"Green Energy & Environment\",\"volume\":\"40 1\",\"pages\":\"\"},\"PeriodicalIF\":14.6000,\"publicationDate\":\"2024-06-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Green Energy & Environment\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://doi.org/10.1016/j.gee.2024.06.007\",\"RegionNum\":1,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Green Energy & Environment","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.1016/j.gee.2024.06.007","RegionNum":1,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
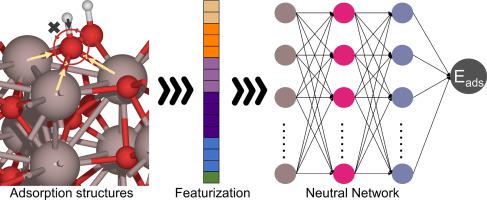
An active learning workflow for predicting hydrogen atom adsorption energies on binary oxides based on local electronic transfer features
Machine learning combined with density functional theory (DFT) enables rapid exploration of catalyst descriptors space such as adsorption energy, facilitating rapid and effective catalyst screening. However, there is still a lack of models for predicting adsorption energies on oxides, due to the complexity of elemental species and the ambiguous coordination environment. This work proposes an active learning workflow (LeNN) founded on local electronic transfer features () and the principle of coordinate rotation invariance. By accurately characterizing the electron transfer to adsorption site atoms and their surrounding geometric structures, LeNN mitigates abrupt feature changes due to different element types and clarifies coordination environments. As a result, it enables the prediction of ∗H adsorption energy on binary oxide surfaces with a mean absolute error (MAE) below 0.18 eV. Moreover, we incorporate local coverage () and leverage neutral network ensemble to establish an active learning workflow, attaining a prediction MAE below 0.2 eV for 5419 multi-∗H adsorption structures. These findings validate the universality and capability of the proposed features in predicting ∗H adsorption energy on binary oxide surfaces.
期刊介绍:
Green Energy & Environment (GEE) is an internationally recognized journal that undergoes a rigorous peer-review process. It focuses on interdisciplinary research related to green energy and the environment, covering a wide range of topics including biofuel and bioenergy, energy storage and networks, catalysis for sustainable processes, and materials for energy and the environment. GEE has a broad scope and encourages the submission of original and innovative research in both fundamental and engineering fields. Additionally, GEE serves as a platform for discussions, summaries, reviews, and previews of the impact of green energy on the eco-environment.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
