Martin Paucar, Daniel Nilsson, Martin Engvall, José Laffita-Mesa, Cilla Söderhäll, Mikael Skorpil, Christer Halldin, Patrik Fazio, Kristina Lagerstedt-Robinson, Göran Solders, Maria Angeria, Andrea Varrone, Mårten Risling, Hong Jiao, Inger Nennesmo, Anna Wedell, Per Svenningsson
{"title":"脊髓小脑共济失调 4 型是由 ZFHX3 基因的 GGC 扩增引起的,与突出的自主神经功能障碍和运动神经元体征有关。","authors":"Martin Paucar, Daniel Nilsson, Martin Engvall, José Laffita-Mesa, Cilla Söderhäll, Mikael Skorpil, Christer Halldin, Patrik Fazio, Kristina Lagerstedt-Robinson, Göran Solders, Maria Angeria, Andrea Varrone, Mårten Risling, Hong Jiao, Inger Nennesmo, Anna Wedell, Per Svenningsson","doi":"10.1111/joim.13815","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Spinocerebellar ataxia 4 (SCA4), characterized in 1996, features adult-onset ataxia, polyneuropathy, and linkage to chromosome 16q22.1; its underlying mutation has remained elusive.</p>\n </section>\n \n <section>\n \n <h3> Objective</h3>\n \n <p>To explore the radiological and neuropathological abnormalities in the entire neuroaxis in SCA4 and search for its mutation.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Three Swedish families with undiagnosed ataxia went through clinical, neurophysiological, and neuroimaging tests, including PET studies and genetic investigations. In four cases, neuropathological assessments of the neuroaxis were performed. Genetic testing included short read whole genome sequencing, short tandem repeat analysis with ExpansionHunter de novo, and long read sequencing.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Novel features for SCA4 include dysautonomia, motor neuron affection, and abnormal eye movements. We found evidence of anticipation; neuroimaging demonstrated atrophy in the cerebellum, brainstem, and spinal cord. [<sup>18</sup>F]FDG-PET demonstrated brain hypometabolism and [<sup>11</sup>C]Flumazenil-PET reduced binding in several brain lobes, insula, thalamus, hypothalamus, and cerebellum. Moderate to severe loss of Purkinje cells in the cerebellum and of motor neurons in the anterior horns of the spinal cord along with pronounced degeneration of posterior tracts was also found. Intranuclear, mainly neuronal, inclusions positive for p62 and ubiquitin were sparse but widespread in the CNS. This finding prompted assessment for nucleotide expansions. A polyglycine stretch encoding GGC expansions in the last exon of the zink finger homeobox 3 gene was identified segregating with disease and not found in 1000 controls.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>SCA4 is a neurodegenerative disease caused by a novel GGC expansion in the coding region of <i>ZFHX3</i>, and its spectrum is expanded to include dysautonomia and neuromuscular manifestations.</p>\n </section>\n </div>","PeriodicalId":196,"journal":{"name":"Journal of Internal Medicine","volume":"296 3","pages":"234-248"},"PeriodicalIF":9.2000,"publicationDate":"2024-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/joim.13815","citationCount":"0","resultStr":"{\"title\":\"Spinocerebellar ataxia type 4 is caused by a GGC expansion in the ZFHX3 gene and is associated with prominent dysautonomia and motor neuron signs\",\"authors\":\"Martin Paucar, Daniel Nilsson, Martin Engvall, José Laffita-Mesa, Cilla Söderhäll, Mikael Skorpil, Christer Halldin, Patrik Fazio, Kristina Lagerstedt-Robinson, Göran Solders, Maria Angeria, Andrea Varrone, Mårten Risling, Hong Jiao, Inger Nennesmo, Anna Wedell, Per Svenningsson\",\"doi\":\"10.1111/joim.13815\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Spinocerebellar ataxia 4 (SCA4), characterized in 1996, features adult-onset ataxia, polyneuropathy, and linkage to chromosome 16q22.1; its underlying mutation has remained elusive.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>To explore the radiological and neuropathological abnormalities in the entire neuroaxis in SCA4 and search for its mutation.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>Three Swedish families with undiagnosed ataxia went through clinical, neurophysiological, and neuroimaging tests, including PET studies and genetic investigations. In four cases, neuropathological assessments of the neuroaxis were performed. Genetic testing included short read whole genome sequencing, short tandem repeat analysis with ExpansionHunter de novo, and long read sequencing.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Novel features for SCA4 include dysautonomia, motor neuron affection, and abnormal eye movements. We found evidence of anticipation; neuroimaging demonstrated atrophy in the cerebellum, brainstem, and spinal cord. [<sup>18</sup>F]FDG-PET demonstrated brain hypometabolism and [<sup>11</sup>C]Flumazenil-PET reduced binding in several brain lobes, insula, thalamus, hypothalamus, and cerebellum. Moderate to severe loss of Purkinje cells in the cerebellum and of motor neurons in the anterior horns of the spinal cord along with pronounced degeneration of posterior tracts was also found. Intranuclear, mainly neuronal, inclusions positive for p62 and ubiquitin were sparse but widespread in the CNS. This finding prompted assessment for nucleotide expansions. A polyglycine stretch encoding GGC expansions in the last exon of the zink finger homeobox 3 gene was identified segregating with disease and not found in 1000 controls.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusions</h3>\\n \\n <p>SCA4 is a neurodegenerative disease caused by a novel GGC expansion in the coding region of <i>ZFHX3</i>, and its spectrum is expanded to include dysautonomia and neuromuscular manifestations.</p>\\n </section>\\n </div>\",\"PeriodicalId\":196,\"journal\":{\"name\":\"Journal of Internal Medicine\",\"volume\":\"296 3\",\"pages\":\"234-248\"},\"PeriodicalIF\":9.2000,\"publicationDate\":\"2024-07-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/joim.13815\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Internal Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/joim.13815\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Internal Medicine","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/joim.13815","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
摘要
背景:脊髓小脑共济失调4(SCA4)于1996年定性,以成人发病的共济失调、多发性神经病为特征,与染色体16q22.1有关联;其潜在的突变一直难以确定:探讨 SCA4 整个神经轴的放射学和神经病理学异常,并寻找其基因突变:三个未确诊共济失调的瑞典家庭接受了临床、神经电生理和神经影像学检查,包括 PET 研究和遗传学调查。在四个病例中,对神经轴进行了神经病理学评估。基因检测包括短读数全基因组测序、短串联重复分析(ExpansionHunter de novo)和长读数测序:结果:SCA4 的新特征包括自主神经功能障碍、运动神经元病变和眼球运动异常。我们发现了预后的证据;神经影像学显示小脑、脑干和脊髓萎缩。[18F]FDG-PET显示大脑代谢低下,[11C]氟马西尼-PET减少了几个脑叶、岛叶、丘脑、下丘脑和小脑的结合。还发现小脑普肯野细胞和脊髓前角运动神经元中度至重度缺失,后束明显退化。核内(主要是神经元)p62 和泛素阳性包涵体稀少,但在中枢神经系统中广泛存在。这一发现促使对核苷酸扩增进行评估。结果发现,在zink finger homeobox 3基因的最后一个外显子中,编码GGC扩增的多甘氨酸伸展与疾病分离,而在1000例对照中没有发现:结论:SCA4 是一种由 ZFHX3 编码区中的新型 GGC 扩增引起的神经退行性疾病,其病谱扩大到包括自主神经功能障碍和神经肌肉表现。
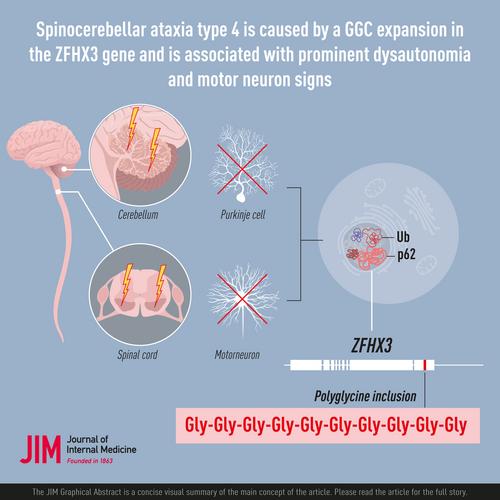
Spinocerebellar ataxia type 4 is caused by a GGC expansion in the ZFHX3 gene and is associated with prominent dysautonomia and motor neuron signs
Background
Spinocerebellar ataxia 4 (SCA4), characterized in 1996, features adult-onset ataxia, polyneuropathy, and linkage to chromosome 16q22.1; its underlying mutation has remained elusive.
Objective
To explore the radiological and neuropathological abnormalities in the entire neuroaxis in SCA4 and search for its mutation.
Methods
Three Swedish families with undiagnosed ataxia went through clinical, neurophysiological, and neuroimaging tests, including PET studies and genetic investigations. In four cases, neuropathological assessments of the neuroaxis were performed. Genetic testing included short read whole genome sequencing, short tandem repeat analysis with ExpansionHunter de novo, and long read sequencing.
Results
Novel features for SCA4 include dysautonomia, motor neuron affection, and abnormal eye movements. We found evidence of anticipation; neuroimaging demonstrated atrophy in the cerebellum, brainstem, and spinal cord. [18F]FDG-PET demonstrated brain hypometabolism and [11C]Flumazenil-PET reduced binding in several brain lobes, insula, thalamus, hypothalamus, and cerebellum. Moderate to severe loss of Purkinje cells in the cerebellum and of motor neurons in the anterior horns of the spinal cord along with pronounced degeneration of posterior tracts was also found. Intranuclear, mainly neuronal, inclusions positive for p62 and ubiquitin were sparse but widespread in the CNS. This finding prompted assessment for nucleotide expansions. A polyglycine stretch encoding GGC expansions in the last exon of the zink finger homeobox 3 gene was identified segregating with disease and not found in 1000 controls.
Conclusions
SCA4 is a neurodegenerative disease caused by a novel GGC expansion in the coding region of ZFHX3, and its spectrum is expanded to include dysautonomia and neuromuscular manifestations.
期刊介绍:
JIM – The Journal of Internal Medicine, in continuous publication since 1863, is an international, peer-reviewed scientific journal. It publishes original work in clinical science, spanning from bench to bedside, encompassing a wide range of internal medicine and its subspecialties. JIM showcases original articles, reviews, brief reports, and research letters in the field of internal medicine.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
