Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Young-Ah Suh, Won-Gun Yun, Wooil Kwon, Jong-Eun Lee, Daeun Kim, Ja-Lok Ku, Jin-Young Jang, Daechan Park
{"title":"定义和追踪胰腺导管腺癌患者来源异种移植模型的亚型。","authors":"Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Young-Ah Suh, Won-Gun Yun, Wooil Kwon, Jong-Eun Lee, Daeun Kim, Ja-Lok Ku, Jin-Young Jang, Daechan Park","doi":"10.1002/cac2.12585","DOIUrl":null,"url":null,"abstract":"<p>Patient-derived xenograft (PDX) models have been used to explore therapeutic opportunities for pancreatic ductal adenocarcinoma (PDAC) [<span>1</span>]. Although original tumor characteristics are altered by cancer-stromal interactions in a PDX-specific manner [<span>2</span>], the implications of clonal evolution from PDAC tumors to PDX are largely unknown.</p><p>In this study, we have conducted a comprehensive genomic analysis using 36 patient-matched PDAC tumor and PDX samples (Figure 1A). The detailed methods regarding this study are described in the Supplementary Materials. The clinical information is summarized in Supplementary Table S1. To compare the somatic mutation profiles of PDAC tumors and PDX, 33 whole exome sequencing data were analyzed by using matched patient blood as a normal control. The proportion of PDX samples with Kirsten Rat Sarcoma Viral Oncogene Homolog (<i>KRAS</i>), Tumor Protein P53 (<i>TP53</i>), Mothers Against Decapentaplegic Homolog 4 (<i>SMAD4</i>), and cyclin-dependent kinase inhibitor 2A (<i>CDKN2A</i>) mutations increased compared to PDAC tumors, indicating that cancerous clones evolved in PDX from primary tumors (Supplementary Table S2-S5, Figure 1B) [<span>3</span>]. Specifically, the frequency of the <i>KRAS</i> G12D mutation increased during PDX establishment, suggesting that this mutation could be responsible for driving clonal evolution in PDX models (Supplementary Table S2). Next, we observed the high correlation of the variant allele frequencies (VAFs) of commonly mutated genes between matched PDAC tumors and PDX in pairwise comparison (Figure 1C), indicating that the overall mutation rate was conserved during PDX construction. When VAFs were compared at the gene level, VAFs of driver genes significantly increased in PDX compared to primary tumors (Figure 1D). Copy number variation (CNV) profiles of protein-coding genes were also similar between the matched samples (Figure 1E, Supplementary Figure S1), while the copy numbers of driver genes became more evident in PDX compared to primary tumors (Figure 1F). Clonality analysis showed that subclones of primary tumors evolved as monoclonal or polyclonal patterns in matched PDX (Supplementary Figure S2). Despite the lack of investigations into clonal evolution over passages, these results suggest that molecular subtypes of PDX could deviate from PDAC tumors via clonal evolution during PDX model construction.</p><p>To investigate whether conventional PDAC subtyping is applicable to PDX, the molecular subtypes defined by Bailey <i>et al.</i> [<span>5</span>] were assigned to PDAC tumors and PDX. PDAC tumors were clearly clustered according to the Bailey gene signatures, showing the worst prognosis of patients with the squamous subtype as previously reported (Figure 1G). However, PDX clustering based on the Bailey gene signatures exhibited 61% (22/36) conflicting subtypes between the matched PDAC tumor and PDX samples (Supplementary Table S6). In particular, the proportions of aberrantly differentiated endocrine exocrine (ADEX) (<i>n</i> = 4) and immunogenic (<i>n</i> = 2) subtypes were reduced, and gene expression of these two subtypes were not clearly distinguished (Figure 1H), implicating the influence of stromal transition in PDX. Also, no distinct survival group was observed by subtype, suggesting that the application of human subtypes for PDX may result in incorrect clinical interpretations.</p><p>Since Bailey subtyping was invalid for PDX, we defined three PDX-specific molecular subtypes (Figure 1I, Supplementary Figure S3, Supplementary Table S7). Gene ontology analysis showed that Cluster 1 signature was related to extracellular matrix organization, whereas Clusters 2 and 3 were similar to squamous and pancreatic progenitor, respectively, among the Bailey subtypes (Supplementary Figure S4). Particularly, PDX Cluster 2 exhibited enrichment of gene expression signatures, such as hypoxia, glycolysis, and DNA replication, associated with rapid clonal expansion (Supplementary Figure S5). Although the survival difference based on the three PDX subtypes was not statistically significant, the prognosis of patients with Cluster 2 exhibited a more pronounced distinction (Figure 1I) and early recurrence within six months after surgery (Supplementary Table S8). To compare the similarity of gene signatures between PDX subtypes and human subtypes, we collected 12 different signatures of 3 human lineages, including stroma, basal and classical from previous studies [<span>5, 4, 6</span>], and then conducted gene overlap analysis (Figure 1J). The numbers in the heatmap represent the numbers of shared signature genes between PDX subtypes and human subtypes, whereby Clusters 1, 2, and 3 corresponded to the stroma, basal, and classical lineages, respectively. Based on these results, the nomenclature of PDX subtypes were assigned to three clusters: PDX-stroma, PDX-basal, and PDX-classical, respectively. Intriguingly, ADEX and immunogenic subtypes did not have any overlapping gene signatures with the three PDX subtypes. Deconvolution analysis of bulk RNA sequencing (RNA-seq) data in this study was performed using cell type signatures [<span>7</span>], confirming a distinct microenvironment in PDAC tumors and PDX (Figure 1K, Supplementary Figures S6 and S7). Next, the subtypes of 36 PDX samples were compared according to widely used subtyping methods for humans (Figure 1L). Notably, stromal lineages were relatively inconsistent across the subtyping methods compared to the other lineages. These results indicate that PDX-stroma requires their own novel gene signatures owing to the unique stromal environment in mice. Tracing the subtype transition between patient-matched PDAC tumors and PDX revealed a lineage-crossing subtype transition in PDX (Figure 1M). The discordance between subtype lineages were found in 55% (20/36) of patient-matched samples (Supplementary Table S9). These inconsistent patterns of subtype transition imply the PDAC tumor heterogeneity underlying the molecular subtypes. To understand the underlying properties of PDAC tumors, we investigated changes in the gene expression between subgroups that transitioned to different PDX subtypes from the same Bailey subtypes (Figure 1N). Gene set enrichment analysis (GSEA) was performed using gene signatures of Bailey subtypes as pre-defined gene sets, as depicted in Supplementary Figure S8. The GSEA results demonstrated that regardless of the Bailey subtype of the primary tumor, the squamous gene set was enriched in subgroups of PDAC tumors transitioning to PDX-basal in the PDX model. Likewise, the pancreatic progenitor gene set was enriched in all subgroups, of which the subtypes transitioned to PDX-classical. These results show that the underlying tumor cells with heterogeneity emerged as a lineage-crossing evolution of PDX-adaptive subclones.</p><p>Genetic discrepancies between primary tumors and model systems may be caused not only by clonal evolution but also by differences in tumor cellularity [<span>8, 9</span>]. High tumor cellularities in matched PDX can explain the increase in VAFs and CNVs of the PDAC-associated genes (Supplementary Figure S9). Our PDX model, by implanting a partial primary tumor, might have lesion-derived bias during clonal evolution in PDX due to intra-tumor heterogeneity. To trace spatial bias in clonal evolution within a tissue, multiple PDX construction from a tissue will be an ideal approach. Although ADEX is considered a subtype with acinar cell contamination [<span>10</span>], our results reveal that basal and classical cancer cells resided within ADEX tissues and expanded during PDX construction, leading to subtype transition. PDXs and organoids tend to better preserve the characteristics of original tumors than cell lines. Nevertheless, organoids undergo subtype changes in response to culture media, and PDXs may exhibit changes in an in vivo microenvironment compared to patient tissues.</p><p>To improve the accuracy of interpreting PDX genomics, it is crucial for further studies to incorporate factors such as the time required for PDX establishment and clinical information such as neoadjuvant chemotherapy. Therefore, clonal change and oncogenetic inconsistencies in PDX models should be investigated in a PDAC-PDX-matched manner, and the use of PDX as a preclinical model system requires careful consideration to accurately predict clinical data and drug responsiveness.</p><p>Jin-Young Jang and Daechan Park conceived and designed the study. Youngmin Han, Won-Gun Yun, Wooil Kwon and Jin-Young Jang collected the biospecimens and clinical information. Young-Ah Suh, Ja-Lok Ku and Jong-Eun Lee performed the experiments. Sangyeop Hyun, Jae Yun Moon and Daeun Kim analyzed the NGS data. Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Won-Gun Yun, Daeun Kim, Jin-Young Jang and Daechan Park interpreted the data. Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Jin-Young Jang and Daechan Park wrote the manuscript. All authors reviewed and approved the final version of the manuscript.</p><p>All authors declare no conflict of interest.</p><p>This research was supported by Global - Learning & Academic research institution for Master's ·PhD students, Postdocs (G-LAMP) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (RS-2023-00285390 to Daechan Park), and the NRF grant funded by the Ministry of Science and ICT (RS-2024-00341899 to Daechan 2022R1A2C2011122 to Jin-Young Jang). This research was partially supported by DNA Link, Inc., and there is no conflict of interest.</p><p>This study was conducted with permission from the Institutional Review Board (IRB, SNUH H-1510-048-710) for research ethics. The biospecimens for this study were provided by Seoul National University Hospital (SNUH) Cancer Tissue Bank and the Biobank of SNUH, a member of the Korea Biobank Network. All samples derived from the Cancer Tissue Bank of SNUH were obtained with informed consent under IRB-approved protocols. All animal experiments were approved by the Institutional Animal Care and Use Committee in the Biomedical Research Institute at Seoul National University Hospital (SNUH-IACUC No. 17-0028-C1A8).</p>","PeriodicalId":9495,"journal":{"name":"Cancer Communications","volume":"44 8","pages":"921-925"},"PeriodicalIF":24.9000,"publicationDate":"2024-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12585","citationCount":"0","resultStr":"{\"title\":\"Defining and tracing subtypes of patient-derived xenograft models in pancreatic ductal adenocarcinoma\",\"authors\":\"Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Young-Ah Suh, Won-Gun Yun, Wooil Kwon, Jong-Eun Lee, Daeun Kim, Ja-Lok Ku, Jin-Young Jang, Daechan Park\",\"doi\":\"10.1002/cac2.12585\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Patient-derived xenograft (PDX) models have been used to explore therapeutic opportunities for pancreatic ductal adenocarcinoma (PDAC) [<span>1</span>]. Although original tumor characteristics are altered by cancer-stromal interactions in a PDX-specific manner [<span>2</span>], the implications of clonal evolution from PDAC tumors to PDX are largely unknown.</p><p>In this study, we have conducted a comprehensive genomic analysis using 36 patient-matched PDAC tumor and PDX samples (Figure 1A). The detailed methods regarding this study are described in the Supplementary Materials. The clinical information is summarized in Supplementary Table S1. To compare the somatic mutation profiles of PDAC tumors and PDX, 33 whole exome sequencing data were analyzed by using matched patient blood as a normal control. The proportion of PDX samples with Kirsten Rat Sarcoma Viral Oncogene Homolog (<i>KRAS</i>), Tumor Protein P53 (<i>TP53</i>), Mothers Against Decapentaplegic Homolog 4 (<i>SMAD4</i>), and cyclin-dependent kinase inhibitor 2A (<i>CDKN2A</i>) mutations increased compared to PDAC tumors, indicating that cancerous clones evolved in PDX from primary tumors (Supplementary Table S2-S5, Figure 1B) [<span>3</span>]. Specifically, the frequency of the <i>KRAS</i> G12D mutation increased during PDX establishment, suggesting that this mutation could be responsible for driving clonal evolution in PDX models (Supplementary Table S2). Next, we observed the high correlation of the variant allele frequencies (VAFs) of commonly mutated genes between matched PDAC tumors and PDX in pairwise comparison (Figure 1C), indicating that the overall mutation rate was conserved during PDX construction. When VAFs were compared at the gene level, VAFs of driver genes significantly increased in PDX compared to primary tumors (Figure 1D). Copy number variation (CNV) profiles of protein-coding genes were also similar between the matched samples (Figure 1E, Supplementary Figure S1), while the copy numbers of driver genes became more evident in PDX compared to primary tumors (Figure 1F). Clonality analysis showed that subclones of primary tumors evolved as monoclonal or polyclonal patterns in matched PDX (Supplementary Figure S2). Despite the lack of investigations into clonal evolution over passages, these results suggest that molecular subtypes of PDX could deviate from PDAC tumors via clonal evolution during PDX model construction.</p><p>To investigate whether conventional PDAC subtyping is applicable to PDX, the molecular subtypes defined by Bailey <i>et al.</i> [<span>5</span>] were assigned to PDAC tumors and PDX. PDAC tumors were clearly clustered according to the Bailey gene signatures, showing the worst prognosis of patients with the squamous subtype as previously reported (Figure 1G). However, PDX clustering based on the Bailey gene signatures exhibited 61% (22/36) conflicting subtypes between the matched PDAC tumor and PDX samples (Supplementary Table S6). In particular, the proportions of aberrantly differentiated endocrine exocrine (ADEX) (<i>n</i> = 4) and immunogenic (<i>n</i> = 2) subtypes were reduced, and gene expression of these two subtypes were not clearly distinguished (Figure 1H), implicating the influence of stromal transition in PDX. Also, no distinct survival group was observed by subtype, suggesting that the application of human subtypes for PDX may result in incorrect clinical interpretations.</p><p>Since Bailey subtyping was invalid for PDX, we defined three PDX-specific molecular subtypes (Figure 1I, Supplementary Figure S3, Supplementary Table S7). Gene ontology analysis showed that Cluster 1 signature was related to extracellular matrix organization, whereas Clusters 2 and 3 were similar to squamous and pancreatic progenitor, respectively, among the Bailey subtypes (Supplementary Figure S4). Particularly, PDX Cluster 2 exhibited enrichment of gene expression signatures, such as hypoxia, glycolysis, and DNA replication, associated with rapid clonal expansion (Supplementary Figure S5). Although the survival difference based on the three PDX subtypes was not statistically significant, the prognosis of patients with Cluster 2 exhibited a more pronounced distinction (Figure 1I) and early recurrence within six months after surgery (Supplementary Table S8). To compare the similarity of gene signatures between PDX subtypes and human subtypes, we collected 12 different signatures of 3 human lineages, including stroma, basal and classical from previous studies [<span>5, 4, 6</span>], and then conducted gene overlap analysis (Figure 1J). The numbers in the heatmap represent the numbers of shared signature genes between PDX subtypes and human subtypes, whereby Clusters 1, 2, and 3 corresponded to the stroma, basal, and classical lineages, respectively. Based on these results, the nomenclature of PDX subtypes were assigned to three clusters: PDX-stroma, PDX-basal, and PDX-classical, respectively. Intriguingly, ADEX and immunogenic subtypes did not have any overlapping gene signatures with the three PDX subtypes. Deconvolution analysis of bulk RNA sequencing (RNA-seq) data in this study was performed using cell type signatures [<span>7</span>], confirming a distinct microenvironment in PDAC tumors and PDX (Figure 1K, Supplementary Figures S6 and S7). Next, the subtypes of 36 PDX samples were compared according to widely used subtyping methods for humans (Figure 1L). Notably, stromal lineages were relatively inconsistent across the subtyping methods compared to the other lineages. These results indicate that PDX-stroma requires their own novel gene signatures owing to the unique stromal environment in mice. Tracing the subtype transition between patient-matched PDAC tumors and PDX revealed a lineage-crossing subtype transition in PDX (Figure 1M). The discordance between subtype lineages were found in 55% (20/36) of patient-matched samples (Supplementary Table S9). These inconsistent patterns of subtype transition imply the PDAC tumor heterogeneity underlying the molecular subtypes. To understand the underlying properties of PDAC tumors, we investigated changes in the gene expression between subgroups that transitioned to different PDX subtypes from the same Bailey subtypes (Figure 1N). Gene set enrichment analysis (GSEA) was performed using gene signatures of Bailey subtypes as pre-defined gene sets, as depicted in Supplementary Figure S8. The GSEA results demonstrated that regardless of the Bailey subtype of the primary tumor, the squamous gene set was enriched in subgroups of PDAC tumors transitioning to PDX-basal in the PDX model. Likewise, the pancreatic progenitor gene set was enriched in all subgroups, of which the subtypes transitioned to PDX-classical. These results show that the underlying tumor cells with heterogeneity emerged as a lineage-crossing evolution of PDX-adaptive subclones.</p><p>Genetic discrepancies between primary tumors and model systems may be caused not only by clonal evolution but also by differences in tumor cellularity [<span>8, 9</span>]. High tumor cellularities in matched PDX can explain the increase in VAFs and CNVs of the PDAC-associated genes (Supplementary Figure S9). Our PDX model, by implanting a partial primary tumor, might have lesion-derived bias during clonal evolution in PDX due to intra-tumor heterogeneity. To trace spatial bias in clonal evolution within a tissue, multiple PDX construction from a tissue will be an ideal approach. Although ADEX is considered a subtype with acinar cell contamination [<span>10</span>], our results reveal that basal and classical cancer cells resided within ADEX tissues and expanded during PDX construction, leading to subtype transition. PDXs and organoids tend to better preserve the characteristics of original tumors than cell lines. Nevertheless, organoids undergo subtype changes in response to culture media, and PDXs may exhibit changes in an in vivo microenvironment compared to patient tissues.</p><p>To improve the accuracy of interpreting PDX genomics, it is crucial for further studies to incorporate factors such as the time required for PDX establishment and clinical information such as neoadjuvant chemotherapy. Therefore, clonal change and oncogenetic inconsistencies in PDX models should be investigated in a PDAC-PDX-matched manner, and the use of PDX as a preclinical model system requires careful consideration to accurately predict clinical data and drug responsiveness.</p><p>Jin-Young Jang and Daechan Park conceived and designed the study. Youngmin Han, Won-Gun Yun, Wooil Kwon and Jin-Young Jang collected the biospecimens and clinical information. Young-Ah Suh, Ja-Lok Ku and Jong-Eun Lee performed the experiments. Sangyeop Hyun, Jae Yun Moon and Daeun Kim analyzed the NGS data. Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Won-Gun Yun, Daeun Kim, Jin-Young Jang and Daechan Park interpreted the data. Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Jin-Young Jang and Daechan Park wrote the manuscript. All authors reviewed and approved the final version of the manuscript.</p><p>All authors declare no conflict of interest.</p><p>This research was supported by Global - Learning & Academic research institution for Master's ·PhD students, Postdocs (G-LAMP) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (RS-2023-00285390 to Daechan Park), and the NRF grant funded by the Ministry of Science and ICT (RS-2024-00341899 to Daechan 2022R1A2C2011122 to Jin-Young Jang). This research was partially supported by DNA Link, Inc., and there is no conflict of interest.</p><p>This study was conducted with permission from the Institutional Review Board (IRB, SNUH H-1510-048-710) for research ethics. The biospecimens for this study were provided by Seoul National University Hospital (SNUH) Cancer Tissue Bank and the Biobank of SNUH, a member of the Korea Biobank Network. All samples derived from the Cancer Tissue Bank of SNUH were obtained with informed consent under IRB-approved protocols. All animal experiments were approved by the Institutional Animal Care and Use Committee in the Biomedical Research Institute at Seoul National University Hospital (SNUH-IACUC No. 17-0028-C1A8).</p>\",\"PeriodicalId\":9495,\"journal\":{\"name\":\"Cancer Communications\",\"volume\":\"44 8\",\"pages\":\"921-925\"},\"PeriodicalIF\":24.9000,\"publicationDate\":\"2024-07-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12585\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Communications\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12585\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Communications","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12585","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
Defining and tracing subtypes of patient-derived xenograft models in pancreatic ductal adenocarcinoma
Patient-derived xenograft (PDX) models have been used to explore therapeutic opportunities for pancreatic ductal adenocarcinoma (PDAC) [1]. Although original tumor characteristics are altered by cancer-stromal interactions in a PDX-specific manner [2], the implications of clonal evolution from PDAC tumors to PDX are largely unknown.
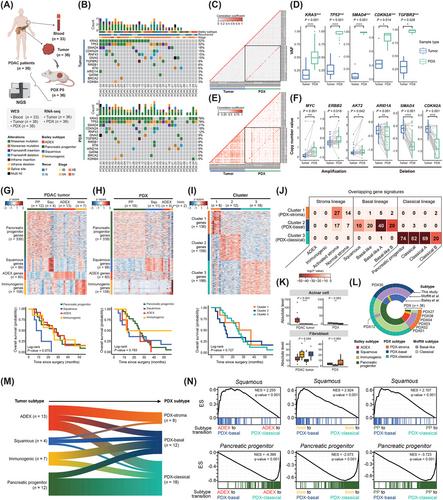
In this study, we have conducted a comprehensive genomic analysis using 36 patient-matched PDAC tumor and PDX samples (Figure 1A). The detailed methods regarding this study are described in the Supplementary Materials. The clinical information is summarized in Supplementary Table S1. To compare the somatic mutation profiles of PDAC tumors and PDX, 33 whole exome sequencing data were analyzed by using matched patient blood as a normal control. The proportion of PDX samples with Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS), Tumor Protein P53 (TP53), Mothers Against Decapentaplegic Homolog 4 (SMAD4), and cyclin-dependent kinase inhibitor 2A (CDKN2A) mutations increased compared to PDAC tumors, indicating that cancerous clones evolved in PDX from primary tumors (Supplementary Table S2-S5, Figure 1B) [3]. Specifically, the frequency of the KRAS G12D mutation increased during PDX establishment, suggesting that this mutation could be responsible for driving clonal evolution in PDX models (Supplementary Table S2). Next, we observed the high correlation of the variant allele frequencies (VAFs) of commonly mutated genes between matched PDAC tumors and PDX in pairwise comparison (Figure 1C), indicating that the overall mutation rate was conserved during PDX construction. When VAFs were compared at the gene level, VAFs of driver genes significantly increased in PDX compared to primary tumors (Figure 1D). Copy number variation (CNV) profiles of protein-coding genes were also similar between the matched samples (Figure 1E, Supplementary Figure S1), while the copy numbers of driver genes became more evident in PDX compared to primary tumors (Figure 1F). Clonality analysis showed that subclones of primary tumors evolved as monoclonal or polyclonal patterns in matched PDX (Supplementary Figure S2). Despite the lack of investigations into clonal evolution over passages, these results suggest that molecular subtypes of PDX could deviate from PDAC tumors via clonal evolution during PDX model construction.
To investigate whether conventional PDAC subtyping is applicable to PDX, the molecular subtypes defined by Bailey et al. [5] were assigned to PDAC tumors and PDX. PDAC tumors were clearly clustered according to the Bailey gene signatures, showing the worst prognosis of patients with the squamous subtype as previously reported (Figure 1G). However, PDX clustering based on the Bailey gene signatures exhibited 61% (22/36) conflicting subtypes between the matched PDAC tumor and PDX samples (Supplementary Table S6). In particular, the proportions of aberrantly differentiated endocrine exocrine (ADEX) (n = 4) and immunogenic (n = 2) subtypes were reduced, and gene expression of these two subtypes were not clearly distinguished (Figure 1H), implicating the influence of stromal transition in PDX. Also, no distinct survival group was observed by subtype, suggesting that the application of human subtypes for PDX may result in incorrect clinical interpretations.
Since Bailey subtyping was invalid for PDX, we defined three PDX-specific molecular subtypes (Figure 1I, Supplementary Figure S3, Supplementary Table S7). Gene ontology analysis showed that Cluster 1 signature was related to extracellular matrix organization, whereas Clusters 2 and 3 were similar to squamous and pancreatic progenitor, respectively, among the Bailey subtypes (Supplementary Figure S4). Particularly, PDX Cluster 2 exhibited enrichment of gene expression signatures, such as hypoxia, glycolysis, and DNA replication, associated with rapid clonal expansion (Supplementary Figure S5). Although the survival difference based on the three PDX subtypes was not statistically significant, the prognosis of patients with Cluster 2 exhibited a more pronounced distinction (Figure 1I) and early recurrence within six months after surgery (Supplementary Table S8). To compare the similarity of gene signatures between PDX subtypes and human subtypes, we collected 12 different signatures of 3 human lineages, including stroma, basal and classical from previous studies [5, 4, 6], and then conducted gene overlap analysis (Figure 1J). The numbers in the heatmap represent the numbers of shared signature genes between PDX subtypes and human subtypes, whereby Clusters 1, 2, and 3 corresponded to the stroma, basal, and classical lineages, respectively. Based on these results, the nomenclature of PDX subtypes were assigned to three clusters: PDX-stroma, PDX-basal, and PDX-classical, respectively. Intriguingly, ADEX and immunogenic subtypes did not have any overlapping gene signatures with the three PDX subtypes. Deconvolution analysis of bulk RNA sequencing (RNA-seq) data in this study was performed using cell type signatures [7], confirming a distinct microenvironment in PDAC tumors and PDX (Figure 1K, Supplementary Figures S6 and S7). Next, the subtypes of 36 PDX samples were compared according to widely used subtyping methods for humans (Figure 1L). Notably, stromal lineages were relatively inconsistent across the subtyping methods compared to the other lineages. These results indicate that PDX-stroma requires their own novel gene signatures owing to the unique stromal environment in mice. Tracing the subtype transition between patient-matched PDAC tumors and PDX revealed a lineage-crossing subtype transition in PDX (Figure 1M). The discordance between subtype lineages were found in 55% (20/36) of patient-matched samples (Supplementary Table S9). These inconsistent patterns of subtype transition imply the PDAC tumor heterogeneity underlying the molecular subtypes. To understand the underlying properties of PDAC tumors, we investigated changes in the gene expression between subgroups that transitioned to different PDX subtypes from the same Bailey subtypes (Figure 1N). Gene set enrichment analysis (GSEA) was performed using gene signatures of Bailey subtypes as pre-defined gene sets, as depicted in Supplementary Figure S8. The GSEA results demonstrated that regardless of the Bailey subtype of the primary tumor, the squamous gene set was enriched in subgroups of PDAC tumors transitioning to PDX-basal in the PDX model. Likewise, the pancreatic progenitor gene set was enriched in all subgroups, of which the subtypes transitioned to PDX-classical. These results show that the underlying tumor cells with heterogeneity emerged as a lineage-crossing evolution of PDX-adaptive subclones.
Genetic discrepancies between primary tumors and model systems may be caused not only by clonal evolution but also by differences in tumor cellularity [8, 9]. High tumor cellularities in matched PDX can explain the increase in VAFs and CNVs of the PDAC-associated genes (Supplementary Figure S9). Our PDX model, by implanting a partial primary tumor, might have lesion-derived bias during clonal evolution in PDX due to intra-tumor heterogeneity. To trace spatial bias in clonal evolution within a tissue, multiple PDX construction from a tissue will be an ideal approach. Although ADEX is considered a subtype with acinar cell contamination [10], our results reveal that basal and classical cancer cells resided within ADEX tissues and expanded during PDX construction, leading to subtype transition. PDXs and organoids tend to better preserve the characteristics of original tumors than cell lines. Nevertheless, organoids undergo subtype changes in response to culture media, and PDXs may exhibit changes in an in vivo microenvironment compared to patient tissues.
To improve the accuracy of interpreting PDX genomics, it is crucial for further studies to incorporate factors such as the time required for PDX establishment and clinical information such as neoadjuvant chemotherapy. Therefore, clonal change and oncogenetic inconsistencies in PDX models should be investigated in a PDAC-PDX-matched manner, and the use of PDX as a preclinical model system requires careful consideration to accurately predict clinical data and drug responsiveness.
Jin-Young Jang and Daechan Park conceived and designed the study. Youngmin Han, Won-Gun Yun, Wooil Kwon and Jin-Young Jang collected the biospecimens and clinical information. Young-Ah Suh, Ja-Lok Ku and Jong-Eun Lee performed the experiments. Sangyeop Hyun, Jae Yun Moon and Daeun Kim analyzed the NGS data. Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Won-Gun Yun, Daeun Kim, Jin-Young Jang and Daechan Park interpreted the data. Sangyeop Hyun, Youngmin Han, Jae Yun Moon, Jin-Young Jang and Daechan Park wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
All authors declare no conflict of interest.
This research was supported by Global - Learning & Academic research institution for Master's ·PhD students, Postdocs (G-LAMP) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (RS-2023-00285390 to Daechan Park), and the NRF grant funded by the Ministry of Science and ICT (RS-2024-00341899 to Daechan 2022R1A2C2011122 to Jin-Young Jang). This research was partially supported by DNA Link, Inc., and there is no conflict of interest.
This study was conducted with permission from the Institutional Review Board (IRB, SNUH H-1510-048-710) for research ethics. The biospecimens for this study were provided by Seoul National University Hospital (SNUH) Cancer Tissue Bank and the Biobank of SNUH, a member of the Korea Biobank Network. All samples derived from the Cancer Tissue Bank of SNUH were obtained with informed consent under IRB-approved protocols. All animal experiments were approved by the Institutional Animal Care and Use Committee in the Biomedical Research Institute at Seoul National University Hospital (SNUH-IACUC No. 17-0028-C1A8).
期刊介绍:
Cancer Communications is an open access, peer-reviewed online journal that encompasses basic, clinical, and translational cancer research. The journal welcomes submissions concerning clinical trials, epidemiology, molecular and cellular biology, and genetics.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
