Fady Baselious, Sebastian Hilscher, Sven Hagemann, Sunita Tripathee, Dina Robaa, Cyril Barinka, Stefan Hüttelmaier, Mike Schutkowski, Wolfgang Sippl
{"title":"利用优化的 AlphaFold 蛋白模型,基于结构设计具有抗神经母细胞瘤活性的选择性 HDAC11 抑制剂。","authors":"Fady Baselious, Sebastian Hilscher, Sven Hagemann, Sunita Tripathee, Dina Robaa, Cyril Barinka, Stefan Hüttelmaier, Mike Schutkowski, Wolfgang Sippl","doi":"10.1002/ardp.202400486","DOIUrl":null,"url":null,"abstract":"<p>AlphaFold is an artificial intelligence approach for predicting the three-dimensional (3D) structures of proteins with atomic accuracy. One challenge that limits the use of AlphaFold models for drug discovery is the correct prediction of folding in the absence of ligands and cofactors, which compromises their direct use. We have previously described the optimization and use of the histone deacetylase 11 (HDAC11) AlphaFold model for the docking of selective inhibitors such as FT895 and SIS17. Based on the predicted binding mode of FT895 in the optimized HDAC11 AlphaFold model, a new scaffold for HDAC11 inhibitors was designed, and the resulting compounds were tested in vitro against various HDAC isoforms. Compound <b>5a</b> proved to be the most active compound with an IC<sub>50</sub> of 365 nM and was able to selectively inhibit HDAC11. Furthermore, docking of <b>5a</b> showed a binding mode comparable to FT895 but could not adopt any reasonable poses in other HDAC isoforms. We further supported the docking results with molecular dynamics simulations that confirmed the predicted binding mode. <b>5a</b> also showed promising activity with an EC<sub>50</sub> of 3.6 µM on neuroblastoma cells.</p>","PeriodicalId":128,"journal":{"name":"Archiv der Pharmazie","volume":"357 10","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-07-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ardp.202400486","citationCount":"0","resultStr":"{\"title\":\"Utilization of an optimized AlphaFold protein model for structure-based design of a selective HDAC11 inhibitor with anti-neuroblastoma activity\",\"authors\":\"Fady Baselious, Sebastian Hilscher, Sven Hagemann, Sunita Tripathee, Dina Robaa, Cyril Barinka, Stefan Hüttelmaier, Mike Schutkowski, Wolfgang Sippl\",\"doi\":\"10.1002/ardp.202400486\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>AlphaFold is an artificial intelligence approach for predicting the three-dimensional (3D) structures of proteins with atomic accuracy. One challenge that limits the use of AlphaFold models for drug discovery is the correct prediction of folding in the absence of ligands and cofactors, which compromises their direct use. We have previously described the optimization and use of the histone deacetylase 11 (HDAC11) AlphaFold model for the docking of selective inhibitors such as FT895 and SIS17. Based on the predicted binding mode of FT895 in the optimized HDAC11 AlphaFold model, a new scaffold for HDAC11 inhibitors was designed, and the resulting compounds were tested in vitro against various HDAC isoforms. Compound <b>5a</b> proved to be the most active compound with an IC<sub>50</sub> of 365 nM and was able to selectively inhibit HDAC11. Furthermore, docking of <b>5a</b> showed a binding mode comparable to FT895 but could not adopt any reasonable poses in other HDAC isoforms. We further supported the docking results with molecular dynamics simulations that confirmed the predicted binding mode. <b>5a</b> also showed promising activity with an EC<sub>50</sub> of 3.6 µM on neuroblastoma cells.</p>\",\"PeriodicalId\":128,\"journal\":{\"name\":\"Archiv der Pharmazie\",\"volume\":\"357 10\",\"pages\":\"\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-07-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ardp.202400486\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Archiv der Pharmazie\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ardp.202400486\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archiv der Pharmazie","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ardp.202400486","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Utilization of an optimized AlphaFold protein model for structure-based design of a selective HDAC11 inhibitor with anti-neuroblastoma activity
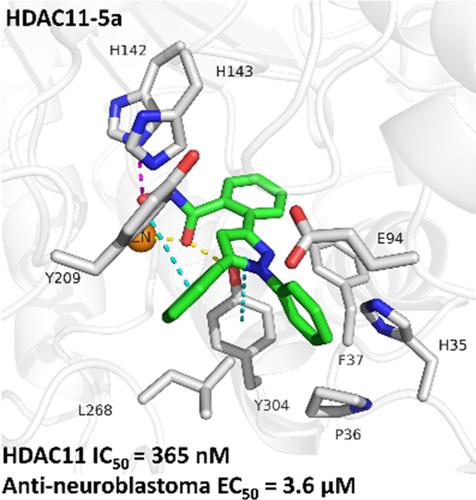
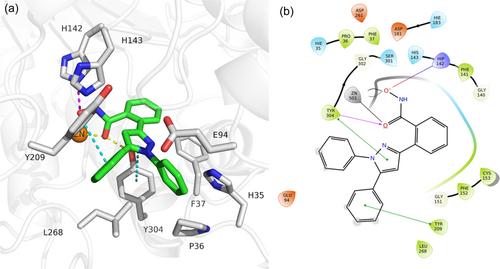
AlphaFold is an artificial intelligence approach for predicting the three-dimensional (3D) structures of proteins with atomic accuracy. One challenge that limits the use of AlphaFold models for drug discovery is the correct prediction of folding in the absence of ligands and cofactors, which compromises their direct use. We have previously described the optimization and use of the histone deacetylase 11 (HDAC11) AlphaFold model for the docking of selective inhibitors such as FT895 and SIS17. Based on the predicted binding mode of FT895 in the optimized HDAC11 AlphaFold model, a new scaffold for HDAC11 inhibitors was designed, and the resulting compounds were tested in vitro against various HDAC isoforms. Compound 5a proved to be the most active compound with an IC50 of 365 nM and was able to selectively inhibit HDAC11. Furthermore, docking of 5a showed a binding mode comparable to FT895 but could not adopt any reasonable poses in other HDAC isoforms. We further supported the docking results with molecular dynamics simulations that confirmed the predicted binding mode. 5a also showed promising activity with an EC50 of 3.6 µM on neuroblastoma cells.
期刊介绍:
Archiv der Pharmazie - Chemistry in Life Sciences is an international journal devoted to research and development in all fields of pharmaceutical and medicinal chemistry. Emphasis is put on papers combining synthetic organic chemistry, structural biology, molecular modelling, bioorganic chemistry, natural products chemistry, biochemistry or analytical methods with pharmaceutical or medicinal aspects such as biological activity. The focus of this journal is put on original research papers, but other scientifically valuable contributions (e.g. reviews, minireviews, highlights, symposia contributions, discussions, and essays) are also welcome.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
