Zheng Wang, Mitsuhiro Kometani, Leonid Zeitlin, Yael Wilnai, Akira Kinoshita, Koh-ichiro Yoshiura, Hiroko Ninomiya, Takeshi Imamura, Long Guo, Jingyi Xue, Li Yan, Hirofumi Ohashi, Yann Pretemer, Shunsuke Kawai, Masaaki Shiina, Kazuhiro Ogata, Daniel H. Cohn, Naomichi Matsumoto, Gen Nishimura, Junya Toguchida, Noriko Miyake, Shiro Ikegawa
{"title":"TGFB2 的潜伏相关肽域的紧身衣区的杂合突变导致卡姆拉蒂-恩格尔曼病 II 型。","authors":"Zheng Wang, Mitsuhiro Kometani, Leonid Zeitlin, Yael Wilnai, Akira Kinoshita, Koh-ichiro Yoshiura, Hiroko Ninomiya, Takeshi Imamura, Long Guo, Jingyi Xue, Li Yan, Hirofumi Ohashi, Yann Pretemer, Shunsuke Kawai, Masaaki Shiina, Kazuhiro Ogata, Daniel H. Cohn, Naomichi Matsumoto, Gen Nishimura, Junya Toguchida, Noriko Miyake, Shiro Ikegawa","doi":"10.1038/s10038-024-01274-1","DOIUrl":null,"url":null,"abstract":"Camurati–Engelmann disease (CED) is an autosomal dominant bone dysplasia characterized by progressive hyperostosis of the skull base and diaphyses of the long bones. CED is further divided into two subtypes, CED1 and CED2, according to the presence or absence of TGFB1 mutations, respectively. In this study, we used exome sequencing to investigate the genetic cause of CED2 in three pedigrees and identified two de novo heterozygous mutations in TGFB2 among the three patients. Both mutations were located in the region of the gene encoding the straitjacket subdomain of the latency-associated peptide (LAP) of pro-TGF-β2. Structural simulations of the mutant LAPs suggested that the mutations could cause significant conformational changes and lead to a reduction in TGF-β2 inactivation. An activity assay confirmed a significant increase in TGF-β2/SMAD signaling. In vitro osteogenic differentiation experiment using iPS cells from one of the CED2 patients showed significantly enhanced ossification, suggesting that the pathogenic mechanism of CED2 is increased activation of TGF-β2 by loss-of-function of the LAP. These results, in combination with the difference in hyperostosis patterns between CED1 and CED2, suggest distinct functions between TGFB1 and TGFB2 in human skeletal development and homeostasis.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 11","pages":"599-605"},"PeriodicalIF":2.3000,"publicationDate":"2024-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Heterozygous mutations in the straitjacket region of the latency-associated peptide domain of TGFB2 cause Camurati–Engelmann disease type II\",\"authors\":\"Zheng Wang, Mitsuhiro Kometani, Leonid Zeitlin, Yael Wilnai, Akira Kinoshita, Koh-ichiro Yoshiura, Hiroko Ninomiya, Takeshi Imamura, Long Guo, Jingyi Xue, Li Yan, Hirofumi Ohashi, Yann Pretemer, Shunsuke Kawai, Masaaki Shiina, Kazuhiro Ogata, Daniel H. Cohn, Naomichi Matsumoto, Gen Nishimura, Junya Toguchida, Noriko Miyake, Shiro Ikegawa\",\"doi\":\"10.1038/s10038-024-01274-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Camurati–Engelmann disease (CED) is an autosomal dominant bone dysplasia characterized by progressive hyperostosis of the skull base and diaphyses of the long bones. CED is further divided into two subtypes, CED1 and CED2, according to the presence or absence of TGFB1 mutations, respectively. In this study, we used exome sequencing to investigate the genetic cause of CED2 in three pedigrees and identified two de novo heterozygous mutations in TGFB2 among the three patients. Both mutations were located in the region of the gene encoding the straitjacket subdomain of the latency-associated peptide (LAP) of pro-TGF-β2. Structural simulations of the mutant LAPs suggested that the mutations could cause significant conformational changes and lead to a reduction in TGF-β2 inactivation. An activity assay confirmed a significant increase in TGF-β2/SMAD signaling. In vitro osteogenic differentiation experiment using iPS cells from one of the CED2 patients showed significantly enhanced ossification, suggesting that the pathogenic mechanism of CED2 is increased activation of TGF-β2 by loss-of-function of the LAP. These results, in combination with the difference in hyperostosis patterns between CED1 and CED2, suggest distinct functions between TGFB1 and TGFB2 in human skeletal development and homeostasis.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 11\",\"pages\":\"599-605\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-07-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01274-1\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01274-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Heterozygous mutations in the straitjacket region of the latency-associated peptide domain of TGFB2 cause Camurati–Engelmann disease type II
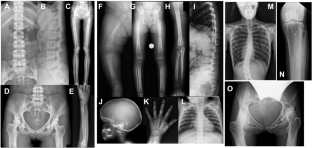
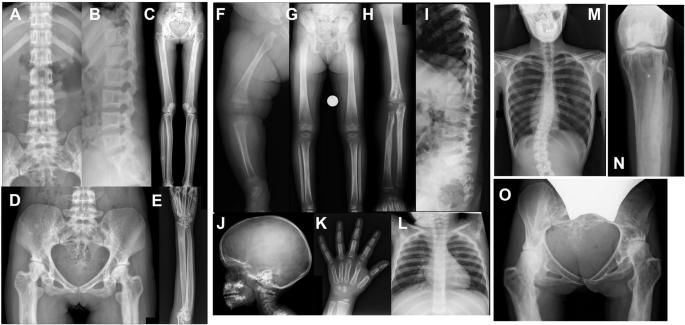
Camurati–Engelmann disease (CED) is an autosomal dominant bone dysplasia characterized by progressive hyperostosis of the skull base and diaphyses of the long bones. CED is further divided into two subtypes, CED1 and CED2, according to the presence or absence of TGFB1 mutations, respectively. In this study, we used exome sequencing to investigate the genetic cause of CED2 in three pedigrees and identified two de novo heterozygous mutations in TGFB2 among the three patients. Both mutations were located in the region of the gene encoding the straitjacket subdomain of the latency-associated peptide (LAP) of pro-TGF-β2. Structural simulations of the mutant LAPs suggested that the mutations could cause significant conformational changes and lead to a reduction in TGF-β2 inactivation. An activity assay confirmed a significant increase in TGF-β2/SMAD signaling. In vitro osteogenic differentiation experiment using iPS cells from one of the CED2 patients showed significantly enhanced ossification, suggesting that the pathogenic mechanism of CED2 is increased activation of TGF-β2 by loss-of-function of the LAP. These results, in combination with the difference in hyperostosis patterns between CED1 and CED2, suggest distinct functions between TGFB1 and TGFB2 in human skeletal development and homeostasis.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
