Mahnoor N Gondal, Saad Ur Rehman Shah, Arul M Chinnaiyan, Marcin Cieslik
{"title":"系统概述单细胞转录组学数据库、其用例和局限性。","authors":"Mahnoor N Gondal, Saad Ur Rehman Shah, Arul M Chinnaiyan, Marcin Cieslik","doi":"10.3389/fbinf.2024.1417428","DOIUrl":null,"url":null,"abstract":"<p><p>Rapid advancements in high-throughput single-cell RNA-seq (scRNA-seq) technologies and experimental protocols have led to the generation of vast amounts of transcriptomic data that populates several online databases and repositories. Here, we systematically examined large-scale scRNA-seq databases, categorizing them based on their scope and purpose such as general, tissue-specific databases, disease-specific databases, cancer-focused databases, and cell type-focused databases. Next, we discuss the technical and methodological challenges associated with curating large-scale scRNA-seq databases, along with current computational solutions. We argue that understanding scRNA-seq databases, including their limitations and assumptions, is crucial for effectively utilizing this data to make robust discoveries and identify novel biological insights. Such platforms can help bridge the gap between computational and wet lab scientists through user-friendly web-based interfaces needed for democratizing access to single-cell data. These platforms would facilitate interdisciplinary research, enabling researchers from various disciplines to collaborate effectively. This review underscores the importance of leveraging computational approaches to unravel the complexities of single-cell data and offers a promising direction for future research in the field.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"4 ","pages":"1417428"},"PeriodicalIF":3.9000,"publicationDate":"2024-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11260681/pdf/","citationCount":"0","resultStr":"{\"title\":\"A systematic overview of single-cell transcriptomics databases, their use cases, and limitations.\",\"authors\":\"Mahnoor N Gondal, Saad Ur Rehman Shah, Arul M Chinnaiyan, Marcin Cieslik\",\"doi\":\"10.3389/fbinf.2024.1417428\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Rapid advancements in high-throughput single-cell RNA-seq (scRNA-seq) technologies and experimental protocols have led to the generation of vast amounts of transcriptomic data that populates several online databases and repositories. Here, we systematically examined large-scale scRNA-seq databases, categorizing them based on their scope and purpose such as general, tissue-specific databases, disease-specific databases, cancer-focused databases, and cell type-focused databases. Next, we discuss the technical and methodological challenges associated with curating large-scale scRNA-seq databases, along with current computational solutions. We argue that understanding scRNA-seq databases, including their limitations and assumptions, is crucial for effectively utilizing this data to make robust discoveries and identify novel biological insights. Such platforms can help bridge the gap between computational and wet lab scientists through user-friendly web-based interfaces needed for democratizing access to single-cell data. These platforms would facilitate interdisciplinary research, enabling researchers from various disciplines to collaborate effectively. This review underscores the importance of leveraging computational approaches to unravel the complexities of single-cell data and offers a promising direction for future research in the field.</p>\",\"PeriodicalId\":73066,\"journal\":{\"name\":\"Frontiers in bioinformatics\",\"volume\":\"4 \",\"pages\":\"1417428\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-07-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11260681/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/fbinf.2024.1417428\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2024.1417428","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
A systematic overview of single-cell transcriptomics databases, their use cases, and limitations.
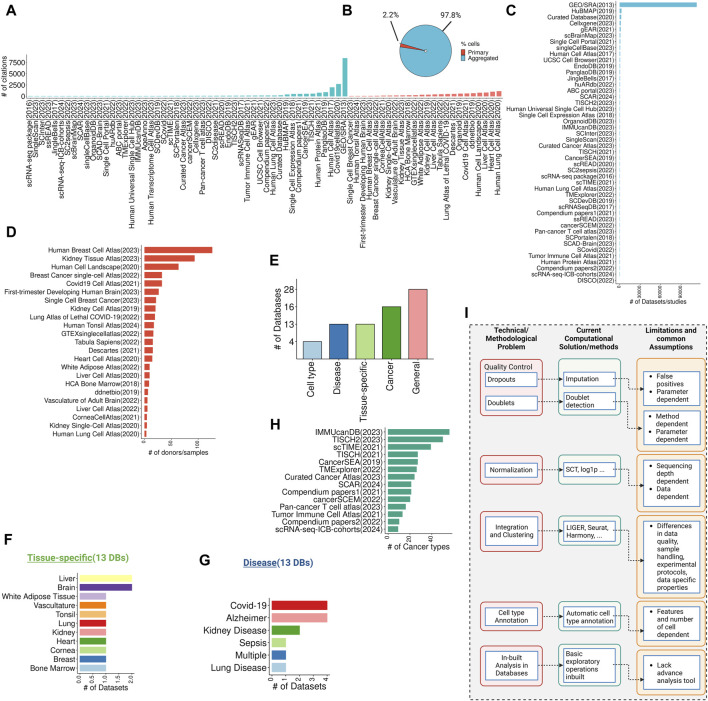
Rapid advancements in high-throughput single-cell RNA-seq (scRNA-seq) technologies and experimental protocols have led to the generation of vast amounts of transcriptomic data that populates several online databases and repositories. Here, we systematically examined large-scale scRNA-seq databases, categorizing them based on their scope and purpose such as general, tissue-specific databases, disease-specific databases, cancer-focused databases, and cell type-focused databases. Next, we discuss the technical and methodological challenges associated with curating large-scale scRNA-seq databases, along with current computational solutions. We argue that understanding scRNA-seq databases, including their limitations and assumptions, is crucial for effectively utilizing this data to make robust discoveries and identify novel biological insights. Such platforms can help bridge the gap between computational and wet lab scientists through user-friendly web-based interfaces needed for democratizing access to single-cell data. These platforms would facilitate interdisciplinary research, enabling researchers from various disciplines to collaborate effectively. This review underscores the importance of leveraging computational approaches to unravel the complexities of single-cell data and offers a promising direction for future research in the field.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
