{"title":"迪乔治综合征在 38 岁时被确诊:低资源环境下的挑战与漏诊的影响》。","authors":"William Kuenstner, Suthee Rapisuwon, Leila Shobab","doi":"10.1210/jcemcr/luae136","DOIUrl":null,"url":null,"abstract":"<p><p>22q11.2 deletion syndrome (22.q11.2 DS) is a genetic syndrome resulting from a microdeletion on chromosome 22. It has a diverse array of manifestations, and most cases are diagnosed early in childhood. We present the case of a 38-year-old female born in a developing country who presented to our clinic to establish care for a history of primary hypothyroidism. She was clinically and biochemically euthyroid on thyroid supplementation. She was also noted to have hypocalcemia in the setting of low PTH, for which the patient was previously prescribed calcitriol. Given a history of cleft palate, abnormal facial features, mild recurrent sinopulmonary infections, and her endocrine history (including short stature with height in the 6th percentile), genetic testing was obtained. She was diagnosed with a heterozygous whole gene deletion of the <i>TBX1</i> gene. Additional genetic evaluation demonstrated a 2.6-Mb microdeleted segment of the 22a11.2 region encompassing 62 genes. The patient was referred to cardiology for evaluation of cardiac involvement given a history of tachyarrhythmia. This case highlights challenges in diagnosis and the implications of a delayed diagnosis of 22.q11.2 DS.</p>","PeriodicalId":73540,"journal":{"name":"JCEM case reports","volume":"2 7","pages":"luae136"},"PeriodicalIF":0.0000,"publicationDate":"2024-07-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11267221/pdf/","citationCount":"0","resultStr":"{\"title\":\"DiGeorge Syndrome Diagnosed at Age 38: Challenges in Low-resource Settings and Implications of a Missed Diagnosis.\",\"authors\":\"William Kuenstner, Suthee Rapisuwon, Leila Shobab\",\"doi\":\"10.1210/jcemcr/luae136\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>22q11.2 deletion syndrome (22.q11.2 DS) is a genetic syndrome resulting from a microdeletion on chromosome 22. It has a diverse array of manifestations, and most cases are diagnosed early in childhood. We present the case of a 38-year-old female born in a developing country who presented to our clinic to establish care for a history of primary hypothyroidism. She was clinically and biochemically euthyroid on thyroid supplementation. She was also noted to have hypocalcemia in the setting of low PTH, for which the patient was previously prescribed calcitriol. Given a history of cleft palate, abnormal facial features, mild recurrent sinopulmonary infections, and her endocrine history (including short stature with height in the 6th percentile), genetic testing was obtained. She was diagnosed with a heterozygous whole gene deletion of the <i>TBX1</i> gene. Additional genetic evaluation demonstrated a 2.6-Mb microdeleted segment of the 22a11.2 region encompassing 62 genes. The patient was referred to cardiology for evaluation of cardiac involvement given a history of tachyarrhythmia. This case highlights challenges in diagnosis and the implications of a delayed diagnosis of 22.q11.2 DS.</p>\",\"PeriodicalId\":73540,\"journal\":{\"name\":\"JCEM case reports\",\"volume\":\"2 7\",\"pages\":\"luae136\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-07-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11267221/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JCEM case reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1210/jcemcr/luae136\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCEM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jcemcr/luae136","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
DiGeorge Syndrome Diagnosed at Age 38: Challenges in Low-resource Settings and Implications of a Missed Diagnosis.
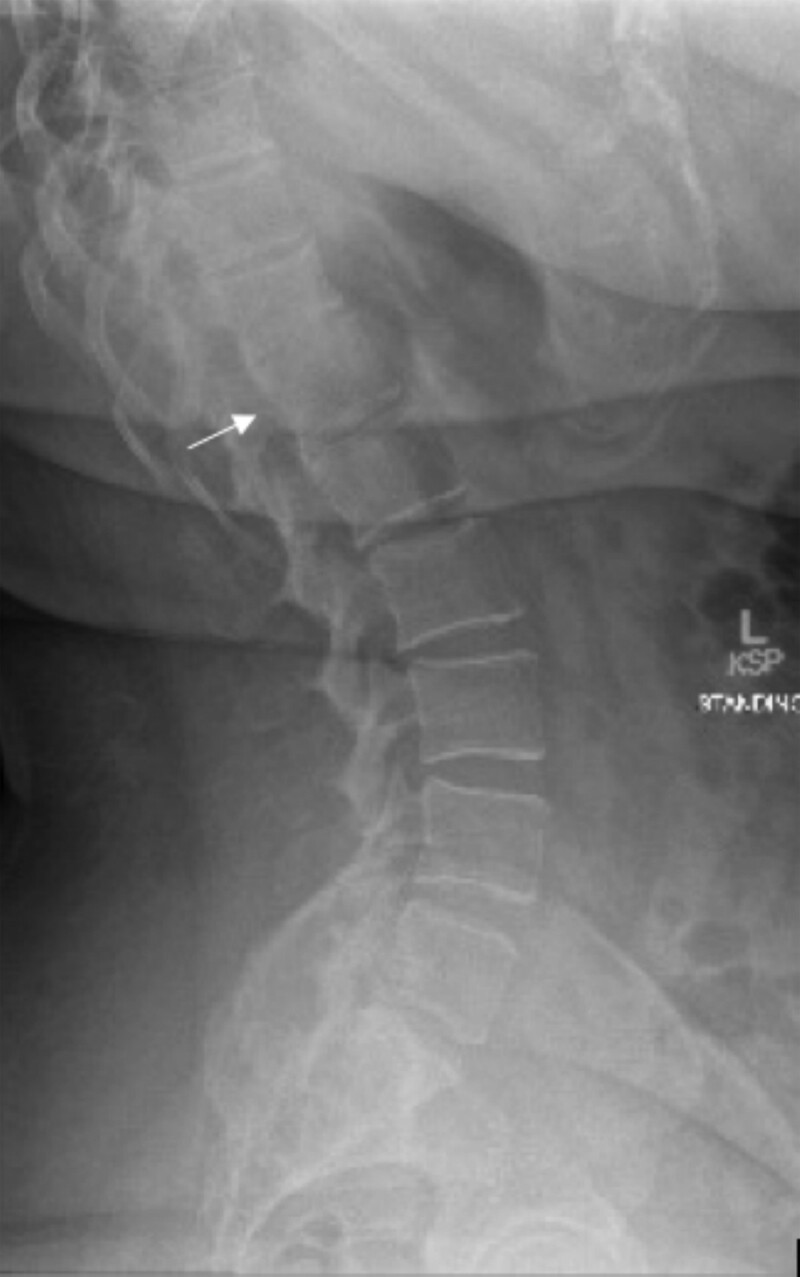
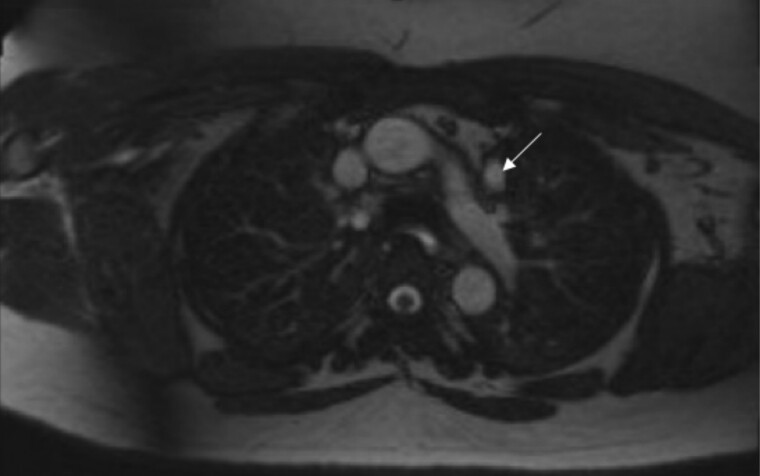
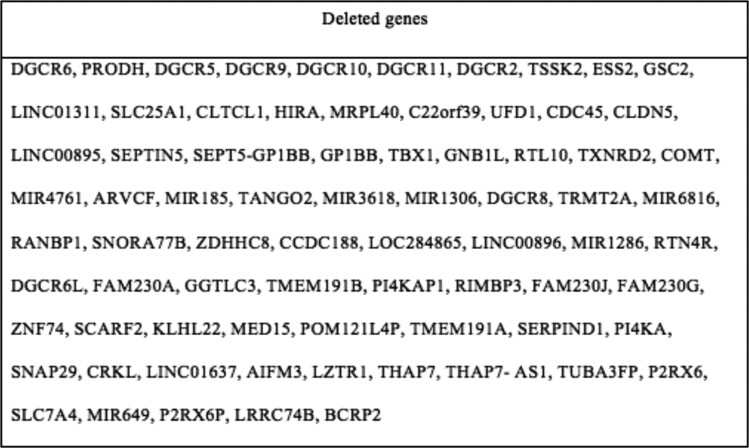
22q11.2 deletion syndrome (22.q11.2 DS) is a genetic syndrome resulting from a microdeletion on chromosome 22. It has a diverse array of manifestations, and most cases are diagnosed early in childhood. We present the case of a 38-year-old female born in a developing country who presented to our clinic to establish care for a history of primary hypothyroidism. She was clinically and biochemically euthyroid on thyroid supplementation. She was also noted to have hypocalcemia in the setting of low PTH, for which the patient was previously prescribed calcitriol. Given a history of cleft palate, abnormal facial features, mild recurrent sinopulmonary infections, and her endocrine history (including short stature with height in the 6th percentile), genetic testing was obtained. She was diagnosed with a heterozygous whole gene deletion of the TBX1 gene. Additional genetic evaluation demonstrated a 2.6-Mb microdeleted segment of the 22a11.2 region encompassing 62 genes. The patient was referred to cardiology for evaluation of cardiac involvement given a history of tachyarrhythmia. This case highlights challenges in diagnosis and the implications of a delayed diagnosis of 22.q11.2 DS.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
