{"title":"同源的患者器官组织揭示了脊髓性肌萎缩症发病初期的神经发育缺陷","authors":"","doi":"10.1016/j.xcrm.2024.101659","DOIUrl":null,"url":null,"abstract":"<p>Whether neurodevelopmental defects underlie postnatal neuronal death in neurodegeneration is an intriguing hypothesis only recently explored. Here, we focus on spinal muscular atrophy (SMA), a neuromuscular disorder caused by reduced survival of motor neuron (SMN) protein levels leading to spinal motor neuron (MN) loss and muscle wasting. Using the first isogenic patient-derived induced pluripotent stem cell (iPSC) model and a spinal cord organoid (SCO) system, we show that SMA SCOs exhibit abnormal morphological development, reduced expression of early neural progenitor markers, and accelerated expression of MN progenitor and MN markers. Longitudinal single-cell RNA sequencing reveals marked defects in neural stem cell specification and fewer MNs, favoring mesodermal progenitors and muscle cells, a bias also seen in early SMA mouse embryos. Surprisingly, <em>SMN2</em>-to-<em>SMN1</em> conversion does not fully reverse these developmental abnormalities. These suggest that early neurodevelopmental defects may underlie later MN degeneration, indicating that postnatal SMN-increasing interventions might not completely amend SMA pathology in all patients.</p>","PeriodicalId":9822,"journal":{"name":"Cell Reports Medicine","volume":"16 1","pages":""},"PeriodicalIF":10.6000,"publicationDate":"2024-07-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Isogenic patient-derived organoids reveal early neurodevelopmental defects in spinal muscular atrophy initiation\",\"authors\":\"\",\"doi\":\"10.1016/j.xcrm.2024.101659\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Whether neurodevelopmental defects underlie postnatal neuronal death in neurodegeneration is an intriguing hypothesis only recently explored. Here, we focus on spinal muscular atrophy (SMA), a neuromuscular disorder caused by reduced survival of motor neuron (SMN) protein levels leading to spinal motor neuron (MN) loss and muscle wasting. Using the first isogenic patient-derived induced pluripotent stem cell (iPSC) model and a spinal cord organoid (SCO) system, we show that SMA SCOs exhibit abnormal morphological development, reduced expression of early neural progenitor markers, and accelerated expression of MN progenitor and MN markers. Longitudinal single-cell RNA sequencing reveals marked defects in neural stem cell specification and fewer MNs, favoring mesodermal progenitors and muscle cells, a bias also seen in early SMA mouse embryos. Surprisingly, <em>SMN2</em>-to-<em>SMN1</em> conversion does not fully reverse these developmental abnormalities. These suggest that early neurodevelopmental defects may underlie later MN degeneration, indicating that postnatal SMN-increasing interventions might not completely amend SMA pathology in all patients.</p>\",\"PeriodicalId\":9822,\"journal\":{\"name\":\"Cell Reports Medicine\",\"volume\":\"16 1\",\"pages\":\"\"},\"PeriodicalIF\":10.6000,\"publicationDate\":\"2024-07-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cell Reports Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1016/j.xcrm.2024.101659\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Reports Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1016/j.xcrm.2024.101659","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Isogenic patient-derived organoids reveal early neurodevelopmental defects in spinal muscular atrophy initiation
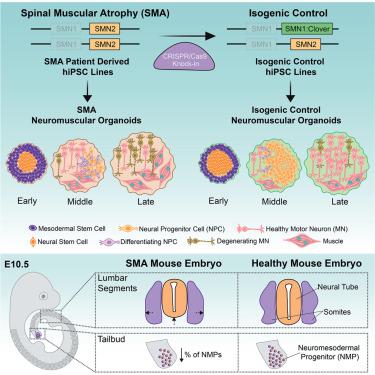
Whether neurodevelopmental defects underlie postnatal neuronal death in neurodegeneration is an intriguing hypothesis only recently explored. Here, we focus on spinal muscular atrophy (SMA), a neuromuscular disorder caused by reduced survival of motor neuron (SMN) protein levels leading to spinal motor neuron (MN) loss and muscle wasting. Using the first isogenic patient-derived induced pluripotent stem cell (iPSC) model and a spinal cord organoid (SCO) system, we show that SMA SCOs exhibit abnormal morphological development, reduced expression of early neural progenitor markers, and accelerated expression of MN progenitor and MN markers. Longitudinal single-cell RNA sequencing reveals marked defects in neural stem cell specification and fewer MNs, favoring mesodermal progenitors and muscle cells, a bias also seen in early SMA mouse embryos. Surprisingly, SMN2-to-SMN1 conversion does not fully reverse these developmental abnormalities. These suggest that early neurodevelopmental defects may underlie later MN degeneration, indicating that postnatal SMN-increasing interventions might not completely amend SMA pathology in all patients.
Cell Reports MedicineBiochemistry, Genetics and Molecular Biology-Biochemistry, Genetics and Molecular Biology (all)
CiteScore
15.00
自引率
1.40%
发文量
231
审稿时长
40 days
期刊介绍:
Cell Reports Medicine is an esteemed open-access journal by Cell Press that publishes groundbreaking research in translational and clinical biomedical sciences, influencing human health and medicine.
Our journal ensures wide visibility and accessibility, reaching scientists and clinicians across various medical disciplines. We publish original research that spans from intriguing human biology concepts to all aspects of clinical work. We encourage submissions that introduce innovative ideas, forging new paths in clinical research and practice. We also welcome studies that provide vital information, enhancing our understanding of current standards of care in diagnosis, treatment, and prognosis. This encompasses translational studies, clinical trials (including long-term follow-ups), genomics, biomarker discovery, and technological advancements that contribute to diagnostics, treatment, and healthcare. Additionally, studies based on vertebrate model organisms are within the scope of the journal, as long as they directly relate to human health and disease.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
