{"title":"LMNB1 基因座的结构变异:解密常染色体显性遗传性成人发病脱髓鞘白营养不良症的病理机制。","authors":"Paola Dimartino PhD, Mariia Zadorozhna PhD, Verónica Yumiceba MSc, Anna Basile MSc, Ilaria Cani MD, Uirá Souto Melo PhD, Jana Henck MSc, Marjolein Breur BSc, Caterina Tonon MD PhD, Raffaele Lodi MD, Alfredo Brusco PhD, Tommaso Pippucci PhD, Foteini-Dionysia Koufi MSc, Elisa Boschetti PhD, Giulia Ramazzotti PhD, Lucia Manzoli MD, Stefano Ratti MD, PhD, Filippo Pinto E Vairo MD, PhD, Martin B. Delatycki MD, PhD, Giovanna Vaula MD, Pietro Cortelli MD, PhD, Marianna Bugiani MD, PhD, Malte Spielmann MD, Elisa Giorgio PhD","doi":"10.1002/ana.27038","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n <h3> Objective</h3>\n \n <p>We aimed to elucidate the pathogenic mechanisms underlying autosomal dominant adult-onset demyelinating leukodystrophy (ADLD), and to understand the genotype/phenotype correlation of structural variants (SVs) in the <i>LMNB1</i> locus.</p>\n </section>\n \n <section>\n \n <h3> Background</h3>\n \n <p>Since the discovery of 3D genome architectures and topologically associating domains (TADs), new pathomechanisms have been postulated for SVs, regardless of gene dosage changes. ADLD is a rare genetic disease associated with duplications (classical ADLD) or noncoding deletions (atypical ADLD) in the <i>LMNB1</i> locus.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>High-throughput chromosome conformation capture, RNA sequencing, histopathological analyses of postmortem brain tissues, and clinical and neuroradiological investigations were performed.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>We collected data from >20 families worldwide carrying SVs in the <i>LMNB1</i> locus and reported strong clinical variability, even among patients carrying duplications of the entire <i>LMNB1</i> gene, ranging from classical and atypical ADLD to asymptomatic carriers. We showed that patients with classic ADLD always carried intra-TAD duplications, resulting in a simple gene dose gain. Atypical ADLD was caused by <i>LMNB1</i> forebrain-specific misexpression due to inter-TAD deletions or duplications. The inter-TAD duplication, which extends centromerically and crosses the 2 TAD boundaries, did not cause ADLD. Our results provide evidence that astrocytes are key players in ADLD pathology.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>Our study sheds light on the 3D genome and TAD structural changes associated with SVs in the <i>LMNB1</i> locus, and shows that a duplication encompassing <i>LMNB1</i> is not sufficient per se to diagnose ADLD, thereby strongly affecting genetic counseling. Our study supports breaking TADs as an emerging pathogenic mechanism that should be considered when studying brain diseases. ANN NEUROL 2024;96:855–870</p>\n </section>\n </div>","PeriodicalId":127,"journal":{"name":"Annals of Neurology","volume":"96 5","pages":"855-870"},"PeriodicalIF":7.5000,"publicationDate":"2024-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ana.27038","citationCount":"0","resultStr":"{\"title\":\"Structural Variants at the LMNB1 Locus: Deciphering Pathomechanisms in Autosomal Dominant Adult-Onset Demyelinating Leukodystrophy\",\"authors\":\"Paola Dimartino PhD, Mariia Zadorozhna PhD, Verónica Yumiceba MSc, Anna Basile MSc, Ilaria Cani MD, Uirá Souto Melo PhD, Jana Henck MSc, Marjolein Breur BSc, Caterina Tonon MD PhD, Raffaele Lodi MD, Alfredo Brusco PhD, Tommaso Pippucci PhD, Foteini-Dionysia Koufi MSc, Elisa Boschetti PhD, Giulia Ramazzotti PhD, Lucia Manzoli MD, Stefano Ratti MD, PhD, Filippo Pinto E Vairo MD, PhD, Martin B. Delatycki MD, PhD, Giovanna Vaula MD, Pietro Cortelli MD, PhD, Marianna Bugiani MD, PhD, Malte Spielmann MD, Elisa Giorgio PhD\",\"doi\":\"10.1002/ana.27038\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>We aimed to elucidate the pathogenic mechanisms underlying autosomal dominant adult-onset demyelinating leukodystrophy (ADLD), and to understand the genotype/phenotype correlation of structural variants (SVs) in the <i>LMNB1</i> locus.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Since the discovery of 3D genome architectures and topologically associating domains (TADs), new pathomechanisms have been postulated for SVs, regardless of gene dosage changes. ADLD is a rare genetic disease associated with duplications (classical ADLD) or noncoding deletions (atypical ADLD) in the <i>LMNB1</i> locus.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>High-throughput chromosome conformation capture, RNA sequencing, histopathological analyses of postmortem brain tissues, and clinical and neuroradiological investigations were performed.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>We collected data from >20 families worldwide carrying SVs in the <i>LMNB1</i> locus and reported strong clinical variability, even among patients carrying duplications of the entire <i>LMNB1</i> gene, ranging from classical and atypical ADLD to asymptomatic carriers. We showed that patients with classic ADLD always carried intra-TAD duplications, resulting in a simple gene dose gain. Atypical ADLD was caused by <i>LMNB1</i> forebrain-specific misexpression due to inter-TAD deletions or duplications. The inter-TAD duplication, which extends centromerically and crosses the 2 TAD boundaries, did not cause ADLD. Our results provide evidence that astrocytes are key players in ADLD pathology.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Interpretation</h3>\\n \\n <p>Our study sheds light on the 3D genome and TAD structural changes associated with SVs in the <i>LMNB1</i> locus, and shows that a duplication encompassing <i>LMNB1</i> is not sufficient per se to diagnose ADLD, thereby strongly affecting genetic counseling. Our study supports breaking TADs as an emerging pathogenic mechanism that should be considered when studying brain diseases. ANN NEUROL 2024;96:855–870</p>\\n </section>\\n </div>\",\"PeriodicalId\":127,\"journal\":{\"name\":\"Annals of Neurology\",\"volume\":\"96 5\",\"pages\":\"855-870\"},\"PeriodicalIF\":7.5000,\"publicationDate\":\"2024-07-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ana.27038\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ana.27038\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Neurology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ana.27038","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
研究目的我们旨在阐明常染色体显性成人型脱髓鞘性白质营养不良症(ALD)的致病机制,并了解LMNB1基因座结构变异(SVs)的基因型/表型相关性:背景:自从发现三维基因组结构和拓扑关联域(TADs)以来,人们对SVs提出了新的病理机制,而不管基因剂量的变化如何。ADLD是一种罕见的遗传病,与LMNB1基因座的重复(典型ADLD)或非编码缺失(非典型ADLD)有关。方法:进行高通量染色体构象捕获、RNA测序、死后脑组织的组织病理学分析以及临床和神经放射学调查:我们收集了全球超过20个携带LMNB1基因位点SV的家族的数据,结果显示,即使是携带整个LMNB1基因重复序列的患者,其临床变异性也很强,从典型和非典型ALD到无症状携带者,不一而足。我们的研究表明,典型 ADLD 患者总是携带 TAD 内重复基因,导致简单的基因剂量增加。非典型 ADLD 是由于 TAD 间的缺失或重复导致 LMNB1 前脑特异性表达错误。TAD间的重复在中心向延伸并跨越了2个TAD的边界,但不会导致ALD。我们的研究结果提供了证据,证明星形胶质细胞是ADLD病理学中的关键角色:我们的研究揭示了与 LMNB1 基因座 SV 相关的三维基因组和 TAD 结构变化,并表明包含 LMNB1 的重复本身不足以诊断 ADLD,从而对遗传咨询产生了重大影响。我们的研究支持将TADs断裂作为一种新兴的致病机制,在研究脑部疾病时应加以考虑。ann neurol 2024.
Structural Variants at the LMNB1 Locus: Deciphering Pathomechanisms in Autosomal Dominant Adult-Onset Demyelinating Leukodystrophy
Objective
We aimed to elucidate the pathogenic mechanisms underlying autosomal dominant adult-onset demyelinating leukodystrophy (ADLD), and to understand the genotype/phenotype correlation of structural variants (SVs) in the LMNB1 locus.
Background
Since the discovery of 3D genome architectures and topologically associating domains (TADs), new pathomechanisms have been postulated for SVs, regardless of gene dosage changes. ADLD is a rare genetic disease associated with duplications (classical ADLD) or noncoding deletions (atypical ADLD) in the LMNB1 locus.
Methods
High-throughput chromosome conformation capture, RNA sequencing, histopathological analyses of postmortem brain tissues, and clinical and neuroradiological investigations were performed.
Results
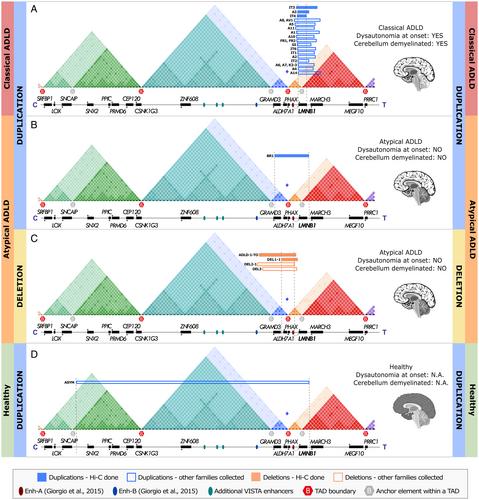
We collected data from >20 families worldwide carrying SVs in the LMNB1 locus and reported strong clinical variability, even among patients carrying duplications of the entire LMNB1 gene, ranging from classical and atypical ADLD to asymptomatic carriers. We showed that patients with classic ADLD always carried intra-TAD duplications, resulting in a simple gene dose gain. Atypical ADLD was caused by LMNB1 forebrain-specific misexpression due to inter-TAD deletions or duplications. The inter-TAD duplication, which extends centromerically and crosses the 2 TAD boundaries, did not cause ADLD. Our results provide evidence that astrocytes are key players in ADLD pathology.
Interpretation
Our study sheds light on the 3D genome and TAD structural changes associated with SVs in the LMNB1 locus, and shows that a duplication encompassing LMNB1 is not sufficient per se to diagnose ADLD, thereby strongly affecting genetic counseling. Our study supports breaking TADs as an emerging pathogenic mechanism that should be considered when studying brain diseases. ANN NEUROL 2024;96:855–870
期刊介绍:
Annals of Neurology publishes original articles with potential for high impact in understanding the pathogenesis, clinical and laboratory features, diagnosis, treatment, outcomes and science underlying diseases of the human nervous system. Articles should ideally be of broad interest to the academic neurological community rather than solely to subspecialists in a particular field. Studies involving experimental model system, including those in cell and organ cultures and animals, of direct translational relevance to the understanding of neurological disease are also encouraged.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
