Johanna Ranta-aho, Kevin J. Felice, Per Harald Jonson, Jaakko Sarparanta, Cédric Yvorel, Ines Harzallah, Renaud Touraine, Lynn Pais, Christina A. Austin-Tse, Vijay S. Ganesh, Melanie C. O'Leary, Heidi L. Rehm, Michael K. Hehir, Sub Subramony, Qian Wu, Bjarne Udd, Marco Savarese
{"title":"蛋白扩展 ACTN2 框移变体通过蛋白聚集导致不同的肌病表型。","authors":"Johanna Ranta-aho, Kevin J. Felice, Per Harald Jonson, Jaakko Sarparanta, Cédric Yvorel, Ines Harzallah, Renaud Touraine, Lynn Pais, Christina A. Austin-Tse, Vijay S. Ganesh, Melanie C. O'Leary, Heidi L. Rehm, Michael K. Hehir, Sub Subramony, Qian Wu, Bjarne Udd, Marco Savarese","doi":"10.1002/acn3.52154","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Objective</h3>\n \n <p>The objective of the study is to characterize the pathomechanisms underlying actininopathies.</p>\n \n <p>Distal myopathies are a group of rare, inherited muscular disorders characterized by progressive loss of muscle fibers that begin in the distal parts of arms and legs. Recently, variants in a new disease gene, <i>ACTN2</i>, have been shown to cause distal myopathy. <i>ACTN2</i>, a gene previously only associated with cardiomyopathies, encodes alpha-actinin-2, a protein expressed in both cardiac and skeletal sarcomeres. The primary function of alpha-actinin-2 is to link actin and titin to the sarcomere Z-disk. New <i>ACTN2</i> variants are continuously discovered; however, the clinical significance of many variants remains unknown. Thus, lack of clear genotype–phenotype correlations in <i>ACTN2</i>-related diseases, actininopathies, persists.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Functional characterization in C2C12 cell model of several <i>ACTN2</i> variants is conducted, including frameshift and missense variants associated with dominant and recessive actininopathies. We assess the genotype–phenotype correlations of actininopathies using clinical data from several patients carrying these variants.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>The results show that the missense variants associated with a recessive form of actininopathy do not cause detectable alpha-actinin-2 aggregates in the cell model. Conversely, dominant frameshift variants causing a protein extension do form alpha-actinin-2 aggregates.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>The results suggest that alpha-actinin-2 aggregation is the disease mechanism underlying some dominant actininopathies, and thus, we recommend that protein-extending frameshift variants in <i>ACTN2</i> should be classified as pathogenic. However, this mechanism is likely elicited by only a limited number of variants. Alternative functional characterization methods should be explored to further investigate other molecular mechanisms underlying actininopathies.</p>\n </section>\n </div>","PeriodicalId":126,"journal":{"name":"Annals of Clinical and Translational Neurology","volume":"11 9","pages":"2392-2405"},"PeriodicalIF":3.9000,"publicationDate":"2024-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/acn3.52154","citationCount":"0","resultStr":"{\"title\":\"Protein-extending ACTN2 frameshift variants cause variable myopathy phenotypes by protein aggregation\",\"authors\":\"Johanna Ranta-aho, Kevin J. Felice, Per Harald Jonson, Jaakko Sarparanta, Cédric Yvorel, Ines Harzallah, Renaud Touraine, Lynn Pais, Christina A. Austin-Tse, Vijay S. Ganesh, Melanie C. O'Leary, Heidi L. Rehm, Michael K. Hehir, Sub Subramony, Qian Wu, Bjarne Udd, Marco Savarese\",\"doi\":\"10.1002/acn3.52154\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>The objective of the study is to characterize the pathomechanisms underlying actininopathies.</p>\\n \\n <p>Distal myopathies are a group of rare, inherited muscular disorders characterized by progressive loss of muscle fibers that begin in the distal parts of arms and legs. Recently, variants in a new disease gene, <i>ACTN2</i>, have been shown to cause distal myopathy. <i>ACTN2</i>, a gene previously only associated with cardiomyopathies, encodes alpha-actinin-2, a protein expressed in both cardiac and skeletal sarcomeres. The primary function of alpha-actinin-2 is to link actin and titin to the sarcomere Z-disk. New <i>ACTN2</i> variants are continuously discovered; however, the clinical significance of many variants remains unknown. Thus, lack of clear genotype–phenotype correlations in <i>ACTN2</i>-related diseases, actininopathies, persists.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>Functional characterization in C2C12 cell model of several <i>ACTN2</i> variants is conducted, including frameshift and missense variants associated with dominant and recessive actininopathies. We assess the genotype–phenotype correlations of actininopathies using clinical data from several patients carrying these variants.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>The results show that the missense variants associated with a recessive form of actininopathy do not cause detectable alpha-actinin-2 aggregates in the cell model. Conversely, dominant frameshift variants causing a protein extension do form alpha-actinin-2 aggregates.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Interpretation</h3>\\n \\n <p>The results suggest that alpha-actinin-2 aggregation is the disease mechanism underlying some dominant actininopathies, and thus, we recommend that protein-extending frameshift variants in <i>ACTN2</i> should be classified as pathogenic. However, this mechanism is likely elicited by only a limited number of variants. Alternative functional characterization methods should be explored to further investigate other molecular mechanisms underlying actininopathies.</p>\\n </section>\\n </div>\",\"PeriodicalId\":126,\"journal\":{\"name\":\"Annals of Clinical and Translational Neurology\",\"volume\":\"11 9\",\"pages\":\"2392-2405\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-08-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/acn3.52154\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Clinical and Translational Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/acn3.52154\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Clinical and Translational Neurology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/acn3.52154","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Protein-extending ACTN2 frameshift variants cause variable myopathy phenotypes by protein aggregation
Objective
The objective of the study is to characterize the pathomechanisms underlying actininopathies.
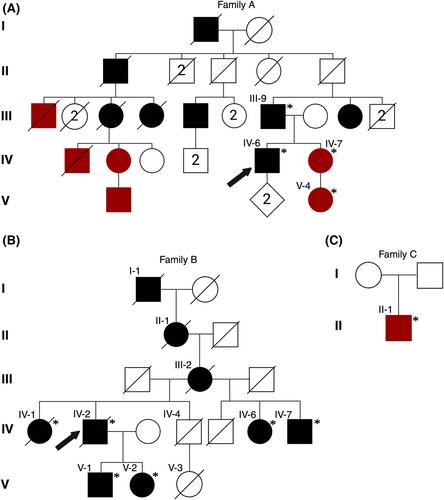
Distal myopathies are a group of rare, inherited muscular disorders characterized by progressive loss of muscle fibers that begin in the distal parts of arms and legs. Recently, variants in a new disease gene, ACTN2, have been shown to cause distal myopathy. ACTN2, a gene previously only associated with cardiomyopathies, encodes alpha-actinin-2, a protein expressed in both cardiac and skeletal sarcomeres. The primary function of alpha-actinin-2 is to link actin and titin to the sarcomere Z-disk. New ACTN2 variants are continuously discovered; however, the clinical significance of many variants remains unknown. Thus, lack of clear genotype–phenotype correlations in ACTN2-related diseases, actininopathies, persists.
Methods
Functional characterization in C2C12 cell model of several ACTN2 variants is conducted, including frameshift and missense variants associated with dominant and recessive actininopathies. We assess the genotype–phenotype correlations of actininopathies using clinical data from several patients carrying these variants.
Results
The results show that the missense variants associated with a recessive form of actininopathy do not cause detectable alpha-actinin-2 aggregates in the cell model. Conversely, dominant frameshift variants causing a protein extension do form alpha-actinin-2 aggregates.
Interpretation
The results suggest that alpha-actinin-2 aggregation is the disease mechanism underlying some dominant actininopathies, and thus, we recommend that protein-extending frameshift variants in ACTN2 should be classified as pathogenic. However, this mechanism is likely elicited by only a limited number of variants. Alternative functional characterization methods should be explored to further investigate other molecular mechanisms underlying actininopathies.
期刊介绍:
Annals of Clinical and Translational Neurology is a peer-reviewed journal for rapid dissemination of high-quality research related to all areas of neurology. The journal publishes original research and scholarly reviews focused on the mechanisms and treatments of diseases of the nervous system; high-impact topics in neurologic education; and other topics of interest to the clinical neuroscience community.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
