{"title":"通过转换器从序列 SNPs 数据中高效推算 HLA。","authors":"Kaho Tanaka, Kosuke Kato, Naoki Nonaka, Jun Seita","doi":"10.1038/s10038-024-01278-x","DOIUrl":null,"url":null,"abstract":"Human leukocyte antigen (HLA) genes are associated with a variety of diseases, yet the direct typing of HLA alleles is both time-consuming and costly. Consequently, various imputation methods leveraging sequential single nucleotide polymorphisms (SNPs) data have been proposed, employing either statistical or deep learning models, such as the convolutional neural network (CNN)-based model, DEEP*HLA. However, these methods exhibit limited imputation efficiency for infrequent alleles and necessitate a large size of reference dataset. In this context, we have developed a Transformer-based model to HLA allele imputation, named “HLA Reliable IMpuatioN by Transformer (HLARIMNT)” designed to exploit the sequential nature of SNPs data. We evaluated HLARIMNT’s performance using two distinct reference panels; Pan-Asian reference panel (n = 530) and Type 1 Diabetes genetics Consortium (T1DGC) reference panel (n = 5225), alongside a combined panel (n = 1060). HLARIMNT demonstrated superior accuracy to DEEP*HLA across several indices, particularly for infrequent alleles. Furthermore, we explored the impact of varying training data sizes on imputation accuracy, finding that HLARIMNT consistently outperformed across all data size. These findings suggest that Transformer-based models can efficiently impute not only HLA types but potentially other gene types from sequential SNPs data.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 10","pages":"533-540"},"PeriodicalIF":2.3000,"publicationDate":"2024-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01278-x.pdf","citationCount":"0","resultStr":"{\"title\":\"Efficient HLA imputation from sequential SNPs data by transformer\",\"authors\":\"Kaho Tanaka, Kosuke Kato, Naoki Nonaka, Jun Seita\",\"doi\":\"10.1038/s10038-024-01278-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Human leukocyte antigen (HLA) genes are associated with a variety of diseases, yet the direct typing of HLA alleles is both time-consuming and costly. Consequently, various imputation methods leveraging sequential single nucleotide polymorphisms (SNPs) data have been proposed, employing either statistical or deep learning models, such as the convolutional neural network (CNN)-based model, DEEP*HLA. However, these methods exhibit limited imputation efficiency for infrequent alleles and necessitate a large size of reference dataset. In this context, we have developed a Transformer-based model to HLA allele imputation, named “HLA Reliable IMpuatioN by Transformer (HLARIMNT)” designed to exploit the sequential nature of SNPs data. We evaluated HLARIMNT’s performance using two distinct reference panels; Pan-Asian reference panel (n = 530) and Type 1 Diabetes genetics Consortium (T1DGC) reference panel (n = 5225), alongside a combined panel (n = 1060). HLARIMNT demonstrated superior accuracy to DEEP*HLA across several indices, particularly for infrequent alleles. Furthermore, we explored the impact of varying training data sizes on imputation accuracy, finding that HLARIMNT consistently outperformed across all data size. These findings suggest that Transformer-based models can efficiently impute not only HLA types but potentially other gene types from sequential SNPs data.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 10\",\"pages\":\"533-540\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-08-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s10038-024-01278-x.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01278-x\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01278-x","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Efficient HLA imputation from sequential SNPs data by transformer
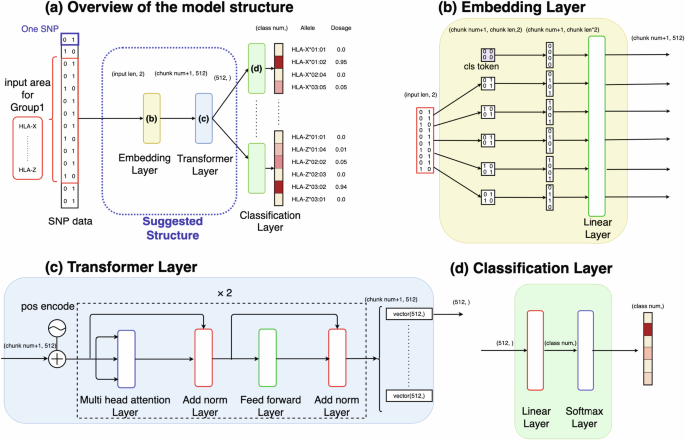
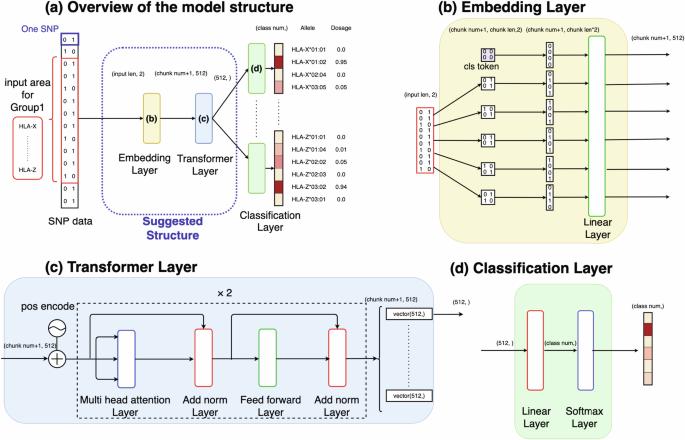
Human leukocyte antigen (HLA) genes are associated with a variety of diseases, yet the direct typing of HLA alleles is both time-consuming and costly. Consequently, various imputation methods leveraging sequential single nucleotide polymorphisms (SNPs) data have been proposed, employing either statistical or deep learning models, such as the convolutional neural network (CNN)-based model, DEEP*HLA. However, these methods exhibit limited imputation efficiency for infrequent alleles and necessitate a large size of reference dataset. In this context, we have developed a Transformer-based model to HLA allele imputation, named “HLA Reliable IMpuatioN by Transformer (HLARIMNT)” designed to exploit the sequential nature of SNPs data. We evaluated HLARIMNT’s performance using two distinct reference panels; Pan-Asian reference panel (n = 530) and Type 1 Diabetes genetics Consortium (T1DGC) reference panel (n = 5225), alongside a combined panel (n = 1060). HLARIMNT demonstrated superior accuracy to DEEP*HLA across several indices, particularly for infrequent alleles. Furthermore, we explored the impact of varying training data sizes on imputation accuracy, finding that HLARIMNT consistently outperformed across all data size. These findings suggest that Transformer-based models can efficiently impute not only HLA types but potentially other gene types from sequential SNPs data.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
