{"title":"用于突变效应预测的蛋白质多级结构特征集成深度学习方法。","authors":"Ai-Ping Pang, Yongsheng Luo, Junping Zhou, Xue Cai, Lianggang Huang, Bo Zhang, Zhi-Qiang Liu, Yu-Guo Zheng","doi":"10.1002/biot.202400203","DOIUrl":null,"url":null,"abstract":"<p>Through iterative rounds of mutation and selection, proteins can be engineered to enhance their desired biological functions. Nevertheless, identifying optimal mutation sites for directed evolution remains challenging due to the vastness of the protein sequence landscape and the epistatic mutational effects across residues. To address this challenge, we introduce MLSmut, a deep learning-based approach that leverages multi-level structural features of proteins. MLSmut extracts salient information from protein co-evolution, sequence semantics, and geometric features to predict the mutational effect. Extensive benchmark evaluations on 10 single-site and two multi-site deep mutation scanning datasets demonstrate that MLSmut surpasses existing methods in predicting mutational outcomes. To overcome the limited training data availability, we employ a two-stage training strategy: initial coarse-tuning on a large corpus of unlabeled protein data followed by fine-tuning on a curated dataset of 40−100 experimental measurements. This approach enables our model to achieve satisfactory performance on downstream protein prediction tasks. Importantly, our model holds the potential to predict the mutational effects of any protein sequence. Collectively, these findings suggest that our approach can substantially reduce the reliance on laborious wet lab experiments and deepen our understanding of the intricate relationships between mutations and protein function.</p>","PeriodicalId":134,"journal":{"name":"Biotechnology Journal","volume":"19 8","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Protein multi-level structure feature-integrated deep learning method for mutational effect prediction\",\"authors\":\"Ai-Ping Pang, Yongsheng Luo, Junping Zhou, Xue Cai, Lianggang Huang, Bo Zhang, Zhi-Qiang Liu, Yu-Guo Zheng\",\"doi\":\"10.1002/biot.202400203\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Through iterative rounds of mutation and selection, proteins can be engineered to enhance their desired biological functions. Nevertheless, identifying optimal mutation sites for directed evolution remains challenging due to the vastness of the protein sequence landscape and the epistatic mutational effects across residues. To address this challenge, we introduce MLSmut, a deep learning-based approach that leverages multi-level structural features of proteins. MLSmut extracts salient information from protein co-evolution, sequence semantics, and geometric features to predict the mutational effect. Extensive benchmark evaluations on 10 single-site and two multi-site deep mutation scanning datasets demonstrate that MLSmut surpasses existing methods in predicting mutational outcomes. To overcome the limited training data availability, we employ a two-stage training strategy: initial coarse-tuning on a large corpus of unlabeled protein data followed by fine-tuning on a curated dataset of 40−100 experimental measurements. This approach enables our model to achieve satisfactory performance on downstream protein prediction tasks. Importantly, our model holds the potential to predict the mutational effects of any protein sequence. Collectively, these findings suggest that our approach can substantially reduce the reliance on laborious wet lab experiments and deepen our understanding of the intricate relationships between mutations and protein function.</p>\",\"PeriodicalId\":134,\"journal\":{\"name\":\"Biotechnology Journal\",\"volume\":\"19 8\",\"pages\":\"\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biotechnology Journal\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/biot.202400203\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biotechnology Journal","FirstCategoryId":"5","ListUrlMain":"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/biot.202400203","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Protein multi-level structure feature-integrated deep learning method for mutational effect prediction
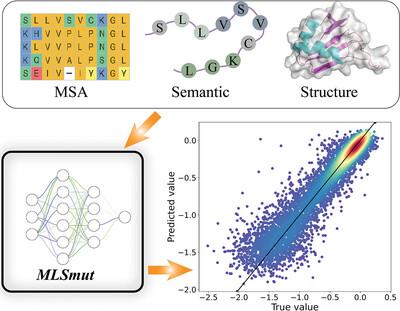
Through iterative rounds of mutation and selection, proteins can be engineered to enhance their desired biological functions. Nevertheless, identifying optimal mutation sites for directed evolution remains challenging due to the vastness of the protein sequence landscape and the epistatic mutational effects across residues. To address this challenge, we introduce MLSmut, a deep learning-based approach that leverages multi-level structural features of proteins. MLSmut extracts salient information from protein co-evolution, sequence semantics, and geometric features to predict the mutational effect. Extensive benchmark evaluations on 10 single-site and two multi-site deep mutation scanning datasets demonstrate that MLSmut surpasses existing methods in predicting mutational outcomes. To overcome the limited training data availability, we employ a two-stage training strategy: initial coarse-tuning on a large corpus of unlabeled protein data followed by fine-tuning on a curated dataset of 40−100 experimental measurements. This approach enables our model to achieve satisfactory performance on downstream protein prediction tasks. Importantly, our model holds the potential to predict the mutational effects of any protein sequence. Collectively, these findings suggest that our approach can substantially reduce the reliance on laborious wet lab experiments and deepen our understanding of the intricate relationships between mutations and protein function.
Biotechnology JournalBiochemistry, Genetics and Molecular Biology-Molecular Medicine
CiteScore
8.90
自引率
2.10%
发文量
123
审稿时长
1.5 months
期刊介绍:
Biotechnology Journal (2019 Journal Citation Reports: 3.543) is fully comprehensive in its scope and publishes strictly peer-reviewed papers covering novel aspects and methods in all areas of biotechnology. Some issues are devoted to a special topic, providing the latest information on the most crucial areas of research and technological advances.
In addition to these special issues, the journal welcomes unsolicited submissions for primary research articles, such as Research Articles, Rapid Communications and Biotech Methods. BTJ also welcomes proposals of Review Articles - please send in a brief outline of the article and the senior author''s CV to the editorial office.
BTJ promotes a special emphasis on:
Systems Biotechnology
Synthetic Biology and Metabolic Engineering
Nanobiotechnology and Biomaterials
Tissue engineering, Regenerative Medicine and Stem cells
Gene Editing, Gene therapy and Immunotherapy
Omics technologies
Industrial Biotechnology, Biopharmaceuticals and Biocatalysis
Bioprocess engineering and Downstream processing
Plant Biotechnology
Biosafety, Biotech Ethics, Science Communication
Methods and Advances.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
