Andrew Leduc, Luke Khoury, Joshua Cantlon, Saad Khan, Nikolai Slavov
{"title":"利用 nPOP 进行大规模并行样品制备,实现多重单细胞蛋白质组学。","authors":"Andrew Leduc, Luke Khoury, Joshua Cantlon, Saad Khan, Nikolai Slavov","doi":"10.1038/s41596-024-01033-8","DOIUrl":null,"url":null,"abstract":"Single-cell proteomics by mass spectrometry (MS) allows the quantification of proteins with high specificity and sensitivity. To increase its throughput, we developed nano-proteomic sample preparation (nPOP), a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides. Here, we describe its protocol with emphasis on its flexibility to prepare samples for different multiplexed MS methods. An implementation using the plexDIA MS multiplexing method, which uses non-isobaric mass tags to barcode peptides from different samples for data-independent acquisition, demonstrates accurate quantification of ~3,000–3,700 proteins per human cell. A separate implementation with isobaric mass tags and prioritized data acquisition demonstrates analysis of 1,827 single cells at a rate of >1,000 single cells per day at a depth of 800–1,200 proteins per human cell. The protocol is implemented by using a cell-dispensing and liquid-handling robot—the CellenONE instrument—and uses readily available consumables, which should facilitate broad adoption. nPOP can be applied to all samples that can be processed to a single-cell suspension. It takes 1 or 2 d to prepare >3,000 single cells. We provide metrics and software (the QuantQC R package) for quality control and data exploration. QuantQC supports the robust scaling of nPOP to higher plex reagents for achieving reliable and scalable single-cell proteomics. nPOP is a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides by using the CellenONE instrument. This protocol describes the liquid handling for multiplexed mass spectrometry proteomics.","PeriodicalId":18901,"journal":{"name":"Nature Protocols","volume":"19 12","pages":"3750-3776"},"PeriodicalIF":16.0000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Massively parallel sample preparation for multiplexed single-cell proteomics using nPOP\",\"authors\":\"Andrew Leduc, Luke Khoury, Joshua Cantlon, Saad Khan, Nikolai Slavov\",\"doi\":\"10.1038/s41596-024-01033-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Single-cell proteomics by mass spectrometry (MS) allows the quantification of proteins with high specificity and sensitivity. To increase its throughput, we developed nano-proteomic sample preparation (nPOP), a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides. Here, we describe its protocol with emphasis on its flexibility to prepare samples for different multiplexed MS methods. An implementation using the plexDIA MS multiplexing method, which uses non-isobaric mass tags to barcode peptides from different samples for data-independent acquisition, demonstrates accurate quantification of ~3,000–3,700 proteins per human cell. A separate implementation with isobaric mass tags and prioritized data acquisition demonstrates analysis of 1,827 single cells at a rate of >1,000 single cells per day at a depth of 800–1,200 proteins per human cell. The protocol is implemented by using a cell-dispensing and liquid-handling robot—the CellenONE instrument—and uses readily available consumables, which should facilitate broad adoption. nPOP can be applied to all samples that can be processed to a single-cell suspension. It takes 1 or 2 d to prepare >3,000 single cells. We provide metrics and software (the QuantQC R package) for quality control and data exploration. QuantQC supports the robust scaling of nPOP to higher plex reagents for achieving reliable and scalable single-cell proteomics. nPOP is a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides by using the CellenONE instrument. This protocol describes the liquid handling for multiplexed mass spectrometry proteomics.\",\"PeriodicalId\":18901,\"journal\":{\"name\":\"Nature Protocols\",\"volume\":\"19 12\",\"pages\":\"3750-3776\"},\"PeriodicalIF\":16.0000,\"publicationDate\":\"2024-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Protocols\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s41596-024-01033-8\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Protocols","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41596-024-01033-8","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Massively parallel sample preparation for multiplexed single-cell proteomics using nPOP
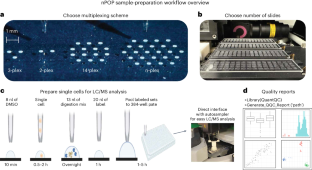
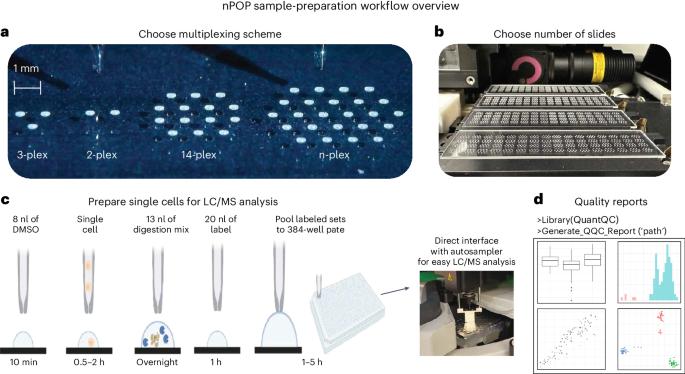
Single-cell proteomics by mass spectrometry (MS) allows the quantification of proteins with high specificity and sensitivity. To increase its throughput, we developed nano-proteomic sample preparation (nPOP), a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides. Here, we describe its protocol with emphasis on its flexibility to prepare samples for different multiplexed MS methods. An implementation using the plexDIA MS multiplexing method, which uses non-isobaric mass tags to barcode peptides from different samples for data-independent acquisition, demonstrates accurate quantification of ~3,000–3,700 proteins per human cell. A separate implementation with isobaric mass tags and prioritized data acquisition demonstrates analysis of 1,827 single cells at a rate of >1,000 single cells per day at a depth of 800–1,200 proteins per human cell. The protocol is implemented by using a cell-dispensing and liquid-handling robot—the CellenONE instrument—and uses readily available consumables, which should facilitate broad adoption. nPOP can be applied to all samples that can be processed to a single-cell suspension. It takes 1 or 2 d to prepare >3,000 single cells. We provide metrics and software (the QuantQC R package) for quality control and data exploration. QuantQC supports the robust scaling of nPOP to higher plex reagents for achieving reliable and scalable single-cell proteomics. nPOP is a method for parallel preparation of thousands of single cells in nanoliter-volume droplets deposited on glass slides by using the CellenONE instrument. This protocol describes the liquid handling for multiplexed mass spectrometry proteomics.
期刊介绍:
Nature Protocols focuses on publishing protocols used to address significant biological and biomedical science research questions, including methods grounded in physics and chemistry with practical applications to biological problems. The journal caters to a primary audience of research scientists and, as such, exclusively publishes protocols with research applications. Protocols primarily aimed at influencing patient management and treatment decisions are not featured.
The specific techniques covered encompass a wide range, including but not limited to: Biochemistry, Cell biology, Cell culture, Chemical modification, Computational biology, Developmental biology, Epigenomics, Genetic analysis, Genetic modification, Genomics, Imaging, Immunology, Isolation, purification, and separation, Lipidomics, Metabolomics, Microbiology, Model organisms, Nanotechnology, Neuroscience, Nucleic-acid-based molecular biology, Pharmacology, Plant biology, Protein analysis, Proteomics, Spectroscopy, Structural biology, Synthetic chemistry, Tissue culture, Toxicology, and Virology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
