Sumeet Patiyal, Palak Tiwari, Mohit Ghai, Aman Dhapola, Anjali Dhall, Gajendra P S Raghava
{"title":"预测转录因子的混合方法。","authors":"Sumeet Patiyal, Palak Tiwari, Mohit Ghai, Aman Dhapola, Anjali Dhall, Gajendra P S Raghava","doi":"10.3389/fbinf.2024.1425419","DOIUrl":null,"url":null,"abstract":"<p><p>Transcription factors are essential DNA-binding proteins that regulate the transcription rate of several genes and control the expression of genes inside a cell. The prediction of transcription factors with high precision is important for understanding biological processes such as cell differentiation, intracellular signaling, and cell-cycle control. In this study, we developed a hybrid method that combines alignment-based and alignment-free methods for predicting transcription factors with higher accuracy. All models have been trained, tested, and evaluated on a large dataset that contains 19,406 transcription factors and 523,560 non-transcription factor protein sequences. To avoid biases in evaluation, the datasets were divided into training and validation/independent datasets, where 80% of the data was used for training, and the remaining 20% was used for external validation. In the case of alignment-free methods, models were developed using machine learning techniques and the composition-based features of a protein. Our best alignment-free model obtained an AUC of 0.97 on an independent dataset. In the case of the alignment-based method, we used BLAST at different cut-offs to predict the transcription factors. Although the alignment-based method demonstrated excellent performance, it was unable to cover all transcription factors due to instances of no hits. To combine the strengths of both methods, we developed a hybrid method that combines alignment-free and alignment-based methods. In the hybrid method, we added the scores of the alignment-free and alignment-based methods and achieved a maximum AUC of 0.99 on the independent dataset. The method proposed in this study performs better than existing methods. We incorporated the best models in the webserver/Python Package Index/standalone package of \"TransFacPred\" (https://webs.iiitd.edu.in/raghava/transfacpred).</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"4 ","pages":"1425419"},"PeriodicalIF":3.9000,"publicationDate":"2024-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11306938/pdf/","citationCount":"0","resultStr":"{\"title\":\"A hybrid approach for predicting transcription factors.\",\"authors\":\"Sumeet Patiyal, Palak Tiwari, Mohit Ghai, Aman Dhapola, Anjali Dhall, Gajendra P S Raghava\",\"doi\":\"10.3389/fbinf.2024.1425419\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Transcription factors are essential DNA-binding proteins that regulate the transcription rate of several genes and control the expression of genes inside a cell. The prediction of transcription factors with high precision is important for understanding biological processes such as cell differentiation, intracellular signaling, and cell-cycle control. In this study, we developed a hybrid method that combines alignment-based and alignment-free methods for predicting transcription factors with higher accuracy. All models have been trained, tested, and evaluated on a large dataset that contains 19,406 transcription factors and 523,560 non-transcription factor protein sequences. To avoid biases in evaluation, the datasets were divided into training and validation/independent datasets, where 80% of the data was used for training, and the remaining 20% was used for external validation. In the case of alignment-free methods, models were developed using machine learning techniques and the composition-based features of a protein. Our best alignment-free model obtained an AUC of 0.97 on an independent dataset. In the case of the alignment-based method, we used BLAST at different cut-offs to predict the transcription factors. Although the alignment-based method demonstrated excellent performance, it was unable to cover all transcription factors due to instances of no hits. To combine the strengths of both methods, we developed a hybrid method that combines alignment-free and alignment-based methods. In the hybrid method, we added the scores of the alignment-free and alignment-based methods and achieved a maximum AUC of 0.99 on the independent dataset. The method proposed in this study performs better than existing methods. We incorporated the best models in the webserver/Python Package Index/standalone package of \\\"TransFacPred\\\" (https://webs.iiitd.edu.in/raghava/transfacpred).</p>\",\"PeriodicalId\":73066,\"journal\":{\"name\":\"Frontiers in bioinformatics\",\"volume\":\"4 \",\"pages\":\"1425419\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11306938/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/fbinf.2024.1425419\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2024.1425419","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
A hybrid approach for predicting transcription factors.
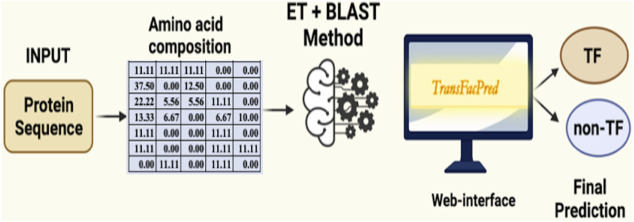
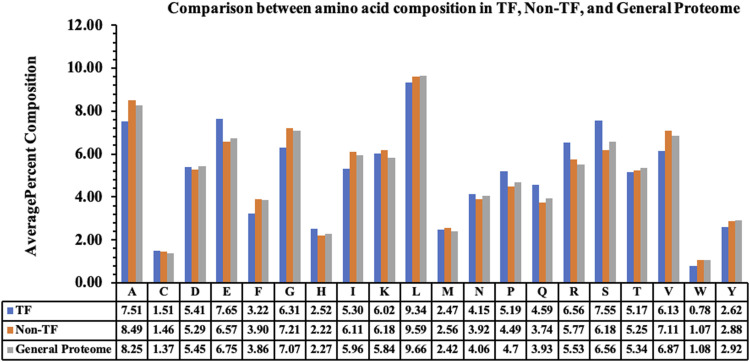
Transcription factors are essential DNA-binding proteins that regulate the transcription rate of several genes and control the expression of genes inside a cell. The prediction of transcription factors with high precision is important for understanding biological processes such as cell differentiation, intracellular signaling, and cell-cycle control. In this study, we developed a hybrid method that combines alignment-based and alignment-free methods for predicting transcription factors with higher accuracy. All models have been trained, tested, and evaluated on a large dataset that contains 19,406 transcription factors and 523,560 non-transcription factor protein sequences. To avoid biases in evaluation, the datasets were divided into training and validation/independent datasets, where 80% of the data was used for training, and the remaining 20% was used for external validation. In the case of alignment-free methods, models were developed using machine learning techniques and the composition-based features of a protein. Our best alignment-free model obtained an AUC of 0.97 on an independent dataset. In the case of the alignment-based method, we used BLAST at different cut-offs to predict the transcription factors. Although the alignment-based method demonstrated excellent performance, it was unable to cover all transcription factors due to instances of no hits. To combine the strengths of both methods, we developed a hybrid method that combines alignment-free and alignment-based methods. In the hybrid method, we added the scores of the alignment-free and alignment-based methods and achieved a maximum AUC of 0.99 on the independent dataset. The method proposed in this study performs better than existing methods. We incorporated the best models in the webserver/Python Package Index/standalone package of "TransFacPred" (https://webs.iiitd.edu.in/raghava/transfacpred).



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
