{"title":"作为 PD-L1 降解剂和 PD-1/PD-L1 相互作用抑制剂的新型 PROTACs 的设计、合成、抗肿瘤活性和机理。","authors":"Feng Zhang , Qimeng Yu , Caiyun Wu , Shishi Sun , Yu Wang , Rui Wang , Zejie Chen , Hua Zhang , Xuqiong Xiong , Annoor Awadasseid , Guowu Rao , Xiaoyin Zhao , Wen Zhang","doi":"10.1016/j.bmc.2024.117867","DOIUrl":null,"url":null,"abstract":"<div><p>Currently, antibody drugs targeting programmed cell death ligand 1 (PD-L1) have achieved promising results in cancer treatment, while the development of small-molecule drugs lags behind. In this study, we designed and synthesized a series of PD-L1-degrading agents based on the PROTAC design principle, utilizing the PD-L1 inhibitor <strong>A56</strong>. Through systematic screening of ligands and linkers and investigating the structure–activity relationship of the degraders, we identified two highly active compounds, <strong>9i</strong> and <strong>9j</strong>. These compounds enhance levels of CD4<sup>+</sup>, CD8<sup>+</sup>, granzyme B, and perforin, demonstrating significant <em>in vivo</em> antitumor effects with a tumor growth inhibition (TGI) of up to 57.35 %. Both compounds facilitate the internalization of PD-L1 from the cell surface and promote its degradation through proteasomal and lysosomal pathways, while also maintaining inhibition of the PD-1/PD-L1 interaction. In summary, our findings provide a novel strategy and mechanism for developing biphenyl-based PROTAC antitumor drugs targeting and degrading PD-L1.</p></div>","PeriodicalId":255,"journal":{"name":"Bioorganic & Medicinal Chemistry","volume":"111 ","pages":"Article 117867"},"PeriodicalIF":3.5000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design, synthesis, anti-tumor activity and mechanism of novel PROTACs as degraders of PD-L1 and inhibitors of PD-1/PD-L1 interaction\",\"authors\":\"Feng Zhang , Qimeng Yu , Caiyun Wu , Shishi Sun , Yu Wang , Rui Wang , Zejie Chen , Hua Zhang , Xuqiong Xiong , Annoor Awadasseid , Guowu Rao , Xiaoyin Zhao , Wen Zhang\",\"doi\":\"10.1016/j.bmc.2024.117867\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Currently, antibody drugs targeting programmed cell death ligand 1 (PD-L1) have achieved promising results in cancer treatment, while the development of small-molecule drugs lags behind. In this study, we designed and synthesized a series of PD-L1-degrading agents based on the PROTAC design principle, utilizing the PD-L1 inhibitor <strong>A56</strong>. Through systematic screening of ligands and linkers and investigating the structure–activity relationship of the degraders, we identified two highly active compounds, <strong>9i</strong> and <strong>9j</strong>. These compounds enhance levels of CD4<sup>+</sup>, CD8<sup>+</sup>, granzyme B, and perforin, demonstrating significant <em>in vivo</em> antitumor effects with a tumor growth inhibition (TGI) of up to 57.35 %. Both compounds facilitate the internalization of PD-L1 from the cell surface and promote its degradation through proteasomal and lysosomal pathways, while also maintaining inhibition of the PD-1/PD-L1 interaction. In summary, our findings provide a novel strategy and mechanism for developing biphenyl-based PROTAC antitumor drugs targeting and degrading PD-L1.</p></div>\",\"PeriodicalId\":255,\"journal\":{\"name\":\"Bioorganic & Medicinal Chemistry\",\"volume\":\"111 \",\"pages\":\"Article 117867\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioorganic & Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0968089624002815\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic & Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0968089624002815","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Design, synthesis, anti-tumor activity and mechanism of novel PROTACs as degraders of PD-L1 and inhibitors of PD-1/PD-L1 interaction
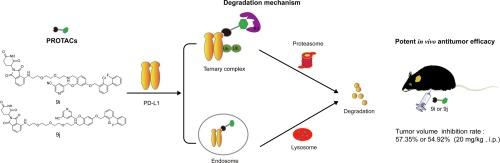
Currently, antibody drugs targeting programmed cell death ligand 1 (PD-L1) have achieved promising results in cancer treatment, while the development of small-molecule drugs lags behind. In this study, we designed and synthesized a series of PD-L1-degrading agents based on the PROTAC design principle, utilizing the PD-L1 inhibitor A56. Through systematic screening of ligands and linkers and investigating the structure–activity relationship of the degraders, we identified two highly active compounds, 9i and 9j. These compounds enhance levels of CD4+, CD8+, granzyme B, and perforin, demonstrating significant in vivo antitumor effects with a tumor growth inhibition (TGI) of up to 57.35 %. Both compounds facilitate the internalization of PD-L1 from the cell surface and promote its degradation through proteasomal and lysosomal pathways, while also maintaining inhibition of the PD-1/PD-L1 interaction. In summary, our findings provide a novel strategy and mechanism for developing biphenyl-based PROTAC antitumor drugs targeting and degrading PD-L1.
期刊介绍:
Bioorganic & Medicinal Chemistry provides an international forum for the publication of full original research papers and critical reviews on molecular interactions in key biological targets such as receptors, channels, enzymes, nucleotides, lipids and saccharides.
The aim of the journal is to promote a better understanding at the molecular level of life processes, and living organisms, as well as the interaction of these with chemical agents. A special feature will be that colour illustrations will be reproduced at no charge to the author, provided that the Editor agrees that colour is essential to the information content of the illustration in question.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
