{"title":"探索无脑畸形谱系的未决病例:整合外显子组和基因组测序以提高诊断率。","authors":"Shogo Furukawa, Mitsuhiro Kato, Akihiko Ishiyama, Tomohiro Kumada, Takeshi Yoshida, Eri Takeshita, Pin Fee Chong, Hideo Yamanouchi, Yuko Kotake, Takayoshi Kyoda, Toshihiro Nomura, Yohane Miyata, Mitsuko Nakashima, Hirotomo Saitsu","doi":"10.1038/s10038-024-01283-0","DOIUrl":null,"url":null,"abstract":"Lissencephaly is a rare brain malformation characterized by abnormal neuronal migration during cortical development. In this study, we performed a comprehensive genetic analysis using next-generation sequencing in 12 unsolved Japanese lissencephaly patients, in whom PAFAH1B1, DCX, TUBA1A, and ARX variants were excluded using the Sanger method. Exome sequencing (ES) was conducted on these 12 patients, identifying pathogenic variants in CEP85L, DYNC1H1, LAMC3, and DCX in four patients. Next, we performed genome sequencing (GS) on eight unsolved patients, and structural variants in PAFAH1B1, including an inversion and microdeletions involving several exons, were detected in three patients. Notably, these microdeletions in PAFAH1B1 could not to be detected by copy number variation (CNV) detection tools based on the depth of coverage methods using ES data. The density of repeat sequences, including Alu sequences or segmental duplications, which increase the susceptibility to structural variations, is very high in some lissencephaly spectrum genes (PAFAH1B1, TUBA1A, DYNC1H1). These missing CNVs were due to the limitations of detecting repeat sequences in ES-based CNV detection tools. Our study suggests that a combined approach integrating ES with GS can contribute to a higher diagnostic yield and a better understanding of the genetic landscape of the lissencephaly spectrum.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 12","pages":"629-637"},"PeriodicalIF":2.6000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring unsolved cases of lissencephaly spectrum: integrating exome and genome sequencing for higher diagnostic yield\",\"authors\":\"Shogo Furukawa, Mitsuhiro Kato, Akihiko Ishiyama, Tomohiro Kumada, Takeshi Yoshida, Eri Takeshita, Pin Fee Chong, Hideo Yamanouchi, Yuko Kotake, Takayoshi Kyoda, Toshihiro Nomura, Yohane Miyata, Mitsuko Nakashima, Hirotomo Saitsu\",\"doi\":\"10.1038/s10038-024-01283-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Lissencephaly is a rare brain malformation characterized by abnormal neuronal migration during cortical development. In this study, we performed a comprehensive genetic analysis using next-generation sequencing in 12 unsolved Japanese lissencephaly patients, in whom PAFAH1B1, DCX, TUBA1A, and ARX variants were excluded using the Sanger method. Exome sequencing (ES) was conducted on these 12 patients, identifying pathogenic variants in CEP85L, DYNC1H1, LAMC3, and DCX in four patients. Next, we performed genome sequencing (GS) on eight unsolved patients, and structural variants in PAFAH1B1, including an inversion and microdeletions involving several exons, were detected in three patients. Notably, these microdeletions in PAFAH1B1 could not to be detected by copy number variation (CNV) detection tools based on the depth of coverage methods using ES data. The density of repeat sequences, including Alu sequences or segmental duplications, which increase the susceptibility to structural variations, is very high in some lissencephaly spectrum genes (PAFAH1B1, TUBA1A, DYNC1H1). These missing CNVs were due to the limitations of detecting repeat sequences in ES-based CNV detection tools. Our study suggests that a combined approach integrating ES with GS can contribute to a higher diagnostic yield and a better understanding of the genetic landscape of the lissencephaly spectrum.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 12\",\"pages\":\"629-637\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2024-08-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01283-0\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01283-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
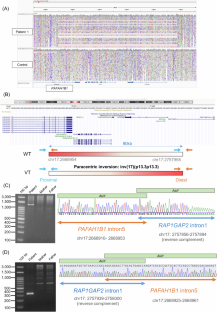
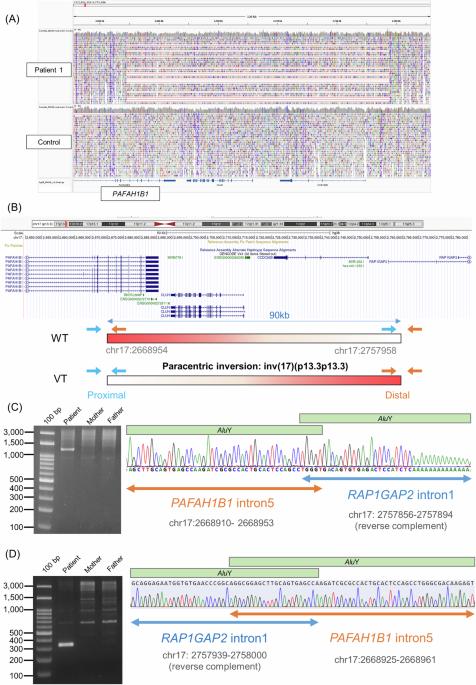
脑裂畸形是一种罕见的脑畸形,其特征是大脑皮层发育过程中神经元迁移异常。在本研究中,我们使用新一代测序技术对 12 例尚未解决的日本脑裂患者进行了全面的遗传分析,其中使用 Sanger 方法排除了 PAFAH1B1、DCX、TUBA1A 和 ARX 变体。我们对这12名患者进行了外显子组测序(ES),在4名患者中发现了CEP85L、DYNC1H1、LAMC3和DCX的致病变异。接下来,我们对 8 例未解决的患者进行了基因组测序(GS),在 3 例患者中检测到 PAFAH1B1 的结构变异,包括倒位和涉及多个外显子的微缺失。值得注意的是,基于ES数据覆盖深度方法的拷贝数变异(CNV)检测工具无法检测到PAFAH1B1中的这些微缺失。在一些脑裂谱基因(PAFAH1B1、TUBA1A、DYNC1H1)中,重复序列(包括 Alu 序列或片段重复)的密度非常高,这增加了结构变异的易感性。这些缺失的 CNV 是由于基于 ES 的 CNV 检测工具在检测重复序列方面的局限性造成的。我们的研究表明,将 ES 与 GS 相结合的方法有助于提高诊断率,并更好地了解无脑畸形谱系的遗传情况。
Exploring unsolved cases of lissencephaly spectrum: integrating exome and genome sequencing for higher diagnostic yield
Lissencephaly is a rare brain malformation characterized by abnormal neuronal migration during cortical development. In this study, we performed a comprehensive genetic analysis using next-generation sequencing in 12 unsolved Japanese lissencephaly patients, in whom PAFAH1B1, DCX, TUBA1A, and ARX variants were excluded using the Sanger method. Exome sequencing (ES) was conducted on these 12 patients, identifying pathogenic variants in CEP85L, DYNC1H1, LAMC3, and DCX in four patients. Next, we performed genome sequencing (GS) on eight unsolved patients, and structural variants in PAFAH1B1, including an inversion and microdeletions involving several exons, were detected in three patients. Notably, these microdeletions in PAFAH1B1 could not to be detected by copy number variation (CNV) detection tools based on the depth of coverage methods using ES data. The density of repeat sequences, including Alu sequences or segmental duplications, which increase the susceptibility to structural variations, is very high in some lissencephaly spectrum genes (PAFAH1B1, TUBA1A, DYNC1H1). These missing CNVs were due to the limitations of detecting repeat sequences in ES-based CNV detection tools. Our study suggests that a combined approach integrating ES with GS can contribute to a higher diagnostic yield and a better understanding of the genetic landscape of the lissencephaly spectrum.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
