Jacob S. Suissa, Gisel Y. De La Cerda, Leland C. Graber, Chloe Jelley, David Wickell, Heather R. Phillips, Ayress D. Grinage, Corrie S. Moreau, Chelsea D. Specht, Jeff J. Doyle, Jacob B. Landis
{"title":"利用 SNP 数据进行系统发生学分析的数据驱动指南","authors":"Jacob S. Suissa, Gisel Y. De La Cerda, Leland C. Graber, Chloe Jelley, David Wickell, Heather R. Phillips, Ayress D. Grinage, Corrie S. Moreau, Chelsea D. Specht, Jeff J. Doyle, Jacob B. Landis","doi":"10.1002/aps3.11611","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Premise</h3>\n \n <p>There is a general lack of consensus on the best practices for filtering of single-nucleotide polymorphisms (SNPs) and whether it is better to use SNPs or include flanking regions (full “locus”) in phylogenomic analyses and subsequent comparative methods.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Using genotyping-by-sequencing data from 22 <i>Glycine</i> species, we assessed the effects of SNP vs. locus usage and SNP retention stringency. We compared branch length, node support, and divergence time estimation across 16 datasets with varying amounts of missing data and total size.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Our results revealed five aspects of phylogenomic data usage that may be generally applicable: (1) tree topology is largely congruent across analyses; (2) filtering strictly for SNP retention (e.g., 90–100%) reduces support and can alter some inferred relationships; (3) absolute branch lengths vary by two orders of magnitude between SNP and locus datasets; (4) data type and branch length variation have little effect on divergence time estimation; and (5) phylograms alter the estimation of ancestral states and rates of morphological evolution.</p>\n </section>\n \n <section>\n \n <h3> Discussion</h3>\n \n <p>Using SNP or locus datasets does not alter phylogenetic inference significantly, unless researchers want or need to use absolute branch lengths. We recommend against using excessive filtering thresholds for SNP retention to reduce the risk of producing inconsistent topologies and generating low support.</p>\n </section>\n </div>","PeriodicalId":8022,"journal":{"name":"Applications in Plant Sciences","volume":"12 6","pages":""},"PeriodicalIF":2.7000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11611","citationCount":"0","resultStr":"{\"title\":\"Data-driven guidelines for phylogenomic analyses using SNP data\",\"authors\":\"Jacob S. Suissa, Gisel Y. De La Cerda, Leland C. Graber, Chloe Jelley, David Wickell, Heather R. Phillips, Ayress D. Grinage, Corrie S. Moreau, Chelsea D. Specht, Jeff J. Doyle, Jacob B. Landis\",\"doi\":\"10.1002/aps3.11611\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Premise</h3>\\n \\n <p>There is a general lack of consensus on the best practices for filtering of single-nucleotide polymorphisms (SNPs) and whether it is better to use SNPs or include flanking regions (full “locus”) in phylogenomic analyses and subsequent comparative methods.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>Using genotyping-by-sequencing data from 22 <i>Glycine</i> species, we assessed the effects of SNP vs. locus usage and SNP retention stringency. We compared branch length, node support, and divergence time estimation across 16 datasets with varying amounts of missing data and total size.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Our results revealed five aspects of phylogenomic data usage that may be generally applicable: (1) tree topology is largely congruent across analyses; (2) filtering strictly for SNP retention (e.g., 90–100%) reduces support and can alter some inferred relationships; (3) absolute branch lengths vary by two orders of magnitude between SNP and locus datasets; (4) data type and branch length variation have little effect on divergence time estimation; and (5) phylograms alter the estimation of ancestral states and rates of morphological evolution.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Discussion</h3>\\n \\n <p>Using SNP or locus datasets does not alter phylogenetic inference significantly, unless researchers want or need to use absolute branch lengths. We recommend against using excessive filtering thresholds for SNP retention to reduce the risk of producing inconsistent topologies and generating low support.</p>\\n </section>\\n </div>\",\"PeriodicalId\":8022,\"journal\":{\"name\":\"Applications in Plant Sciences\",\"volume\":\"12 6\",\"pages\":\"\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-08-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11611\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Applications in Plant Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/aps3.11611\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PLANT SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Applications in Plant Sciences","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/aps3.11611","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
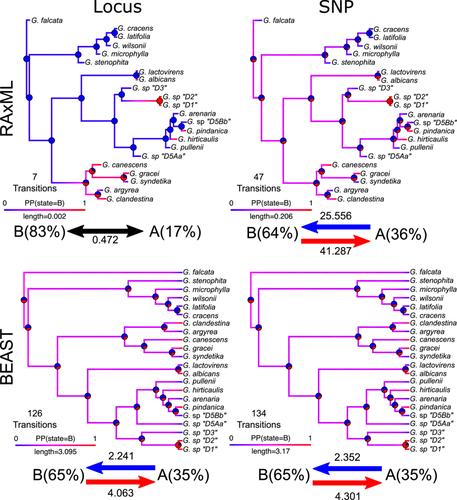
关于过滤单核苷酸多态性(SNP)的最佳方法,以及在系统发生组分析和后续比较方法中使用 SNP 还是包括侧翼区域(完整的 "位点")更好,目前普遍缺乏共识。我们比较了 16 个数据集的分支长度、节点支持率和分化时间估计,这些数据集的缺失数据量和总规模各不相同。我们的结果揭示了系统发生组数据使用的五个方面,这些方面可能具有普遍适用性:(1)树拓扑结构在不同分析中基本一致;(2)严格筛选 SNP 保留率(如 90%-100%)会降低支持率,并且会影响树的分化时间、(3) SNP 数据集和基因座数据集的绝对分支长度相差两个数量级;(4) 数据类型和分支长度的变化对分歧时间的估计影响不大;(5) 系统图会改变对祖先状态和形态进化速率的估计。我们建议不要使用过高的 SNP 保留过滤阈值,以降低产生不一致拓扑和低支持度的风险。
Data-driven guidelines for phylogenomic analyses using SNP data
Premise
There is a general lack of consensus on the best practices for filtering of single-nucleotide polymorphisms (SNPs) and whether it is better to use SNPs or include flanking regions (full “locus”) in phylogenomic analyses and subsequent comparative methods.
Methods
Using genotyping-by-sequencing data from 22 Glycine species, we assessed the effects of SNP vs. locus usage and SNP retention stringency. We compared branch length, node support, and divergence time estimation across 16 datasets with varying amounts of missing data and total size.
Results
Our results revealed five aspects of phylogenomic data usage that may be generally applicable: (1) tree topology is largely congruent across analyses; (2) filtering strictly for SNP retention (e.g., 90–100%) reduces support and can alter some inferred relationships; (3) absolute branch lengths vary by two orders of magnitude between SNP and locus datasets; (4) data type and branch length variation have little effect on divergence time estimation; and (5) phylograms alter the estimation of ancestral states and rates of morphological evolution.
Discussion
Using SNP or locus datasets does not alter phylogenetic inference significantly, unless researchers want or need to use absolute branch lengths. We recommend against using excessive filtering thresholds for SNP retention to reduce the risk of producing inconsistent topologies and generating low support.
期刊介绍:
Applications in Plant Sciences (APPS) is a monthly, peer-reviewed, open access journal promoting the rapid dissemination of newly developed, innovative tools and protocols in all areas of the plant sciences, including genetics, structure, function, development, evolution, systematics, and ecology. Given the rapid progress today in technology and its application in the plant sciences, the goal of APPS is to foster communication within the plant science community to advance scientific research. APPS is a publication of the Botanical Society of America, originating in 2009 as the American Journal of Botany''s online-only section, AJB Primer Notes & Protocols in the Plant Sciences.
APPS publishes the following types of articles: (1) Protocol Notes describe new methods and technological advancements; (2) Genomic Resources Articles characterize the development and demonstrate the usefulness of newly developed genomic resources, including transcriptomes; (3) Software Notes detail new software applications; (4) Application Articles illustrate the application of a new protocol, method, or software application within the context of a larger study; (5) Review Articles evaluate available techniques, methods, or protocols; (6) Primer Notes report novel genetic markers with evidence of wide applicability.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
