{"title":"利用密度泛函理论理解 Cu19 团簇上分子氧和原子氧的 CO 氧化反应","authors":"ANJANI NANDAN PANDEY, RAMAN K. SINGH","doi":"10.1007/s12039-024-02291-5","DOIUrl":null,"url":null,"abstract":"<div><p>In this work, we present the CO oxidation by molecular and atomic oxygen on the Cu<sub>3</sub> site of the Cu<sub>19</sub> cluster employing the density functional theory (DFT)-PW91PW91/[LANL2DZ, 6-31G(d)] level. The computed results demonstrate that the O atom, O<sub>2</sub>, and CO molecule adsorptions on the copper cluster are all chemical. For the CO oxidation by O<sub>2</sub> molecules that leads to the formation of C–O bonds and the dissociation of O–O bonds, the Langmuir–Hinshelwood (LH) mechanism is preferred. On the other hand, the Eley–Rideal (ER) mechanism is slightly favored by the oxidation of CO by atomic oxygen. According to the intrinsic reaction coordinate (IRC) calculation, the activation energy for CO oxidation is 4.02 kcal/mol for molecular oxygen and 3.17 kcal/mol for atomic oxygen. Therefore, molecular and atomic oxygen are very reactive for CO oxidation on the Cu<sub>19</sub> cluster. To check the applicability of the global hardness response (GHR) profile satisfying the maximum hardness principle along the IRC in the metal cluster reactions, the GHR profile for the oxidation reaction of CO with molecular oxygen and atomic oxygen was computed. The results indicate that this meets the principle of maximum hardness, effectively showcasing the use of DFT methods to analyze the global hardness profile using frontier molecular orbital energy in the context of metal cluster reaction pathways.</p><h3>Graphical abstract</h3><p>The CO oxidation by molecular and atomic oxygen on the Cu<sub>3</sub> site of the Cu<sub>19</sub> cluster employing the density functional theory has been studied. The results show that the CO oxidation by atomic oxygen slightly favors the Eley–Rideal (ER) mechanism, while the CO oxidation by molecular oxygen prefers the Langmuir–Hinshelwood (LH) mechanism. Both molecular and atomic oxygen are very reactive for CO oxidation. Furthermore, the global hardness profile along the intrinsic reaction coordinate follows the maximum hardness principle.\n</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":616,"journal":{"name":"Journal of Chemical Sciences","volume":"136 3","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Understanding of CO oxidation reaction by molecular and atomic oxygen on Cu19 cluster using the density functional theory\",\"authors\":\"ANJANI NANDAN PANDEY, RAMAN K. SINGH\",\"doi\":\"10.1007/s12039-024-02291-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>In this work, we present the CO oxidation by molecular and atomic oxygen on the Cu<sub>3</sub> site of the Cu<sub>19</sub> cluster employing the density functional theory (DFT)-PW91PW91/[LANL2DZ, 6-31G(d)] level. The computed results demonstrate that the O atom, O<sub>2</sub>, and CO molecule adsorptions on the copper cluster are all chemical. For the CO oxidation by O<sub>2</sub> molecules that leads to the formation of C–O bonds and the dissociation of O–O bonds, the Langmuir–Hinshelwood (LH) mechanism is preferred. On the other hand, the Eley–Rideal (ER) mechanism is slightly favored by the oxidation of CO by atomic oxygen. According to the intrinsic reaction coordinate (IRC) calculation, the activation energy for CO oxidation is 4.02 kcal/mol for molecular oxygen and 3.17 kcal/mol for atomic oxygen. Therefore, molecular and atomic oxygen are very reactive for CO oxidation on the Cu<sub>19</sub> cluster. To check the applicability of the global hardness response (GHR) profile satisfying the maximum hardness principle along the IRC in the metal cluster reactions, the GHR profile for the oxidation reaction of CO with molecular oxygen and atomic oxygen was computed. The results indicate that this meets the principle of maximum hardness, effectively showcasing the use of DFT methods to analyze the global hardness profile using frontier molecular orbital energy in the context of metal cluster reaction pathways.</p><h3>Graphical abstract</h3><p>The CO oxidation by molecular and atomic oxygen on the Cu<sub>3</sub> site of the Cu<sub>19</sub> cluster employing the density functional theory has been studied. The results show that the CO oxidation by atomic oxygen slightly favors the Eley–Rideal (ER) mechanism, while the CO oxidation by molecular oxygen prefers the Langmuir–Hinshelwood (LH) mechanism. Both molecular and atomic oxygen are very reactive for CO oxidation. Furthermore, the global hardness profile along the intrinsic reaction coordinate follows the maximum hardness principle.\\n</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":616,\"journal\":{\"name\":\"Journal of Chemical Sciences\",\"volume\":\"136 3\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-08-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Sciences\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s12039-024-02291-5\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Sciences","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s12039-024-02291-5","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
在这项工作中,我们采用密度泛函理论(DFT)-PW91PW91/[LANL2DZ, 6-31G(d)]水平,研究了分子氧和原子氧在 Cu19 团簇的 Cu3 位点上的 CO 氧化作用。计算结果表明,铜簇上的 O 原子、O2 和 CO 分子吸附都是化学吸附。对于 CO 被 O2 分子氧化导致 C-O 键的形成和 O-O 键的解离,Langmuir-Hinshelwood(LH)机制是首选机制。另一方面,CO 被原子氧氧化后,Eley-Rideal(ER)机理略占优势。根据本征反应坐标(IRC)计算,分子氧氧化 CO 的活化能为 4.02 kcal/mol,原子氧氧化 CO 的活化能为 3.17 kcal/mol。因此,分子氧和原子氧对 Cu19 团簇上的 CO 氧化反应非常活跃。为了检验符合沿 IRC 最大硬度原则的全局硬度响应(GHR)曲线在金属簇反应中的适用性,计算了 CO 与分子氧和原子氧氧化反应的 GHR 曲线。结果表明,这符合最大硬度原则,有效地展示了在金属簇反应途径中使用前沿分子轨道能的 DFT 方法来分析全局硬度曲线。结果表明,原子氧对 CO 的氧化作用略微倾向于 Eley-Rideal(ER)机理,而分子氧对 CO 的氧化作用则倾向于 Langmuir-Hinshelwood(LH)机理。分子氧和原子氧对一氧化碳的氧化都非常活跃。此外,沿固有反应坐标的总体硬度曲线遵循最大硬度原则。
Understanding of CO oxidation reaction by molecular and atomic oxygen on Cu19 cluster using the density functional theory
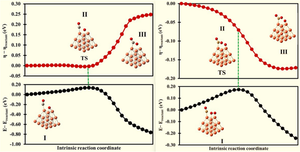
In this work, we present the CO oxidation by molecular and atomic oxygen on the Cu3 site of the Cu19 cluster employing the density functional theory (DFT)-PW91PW91/[LANL2DZ, 6-31G(d)] level. The computed results demonstrate that the O atom, O2, and CO molecule adsorptions on the copper cluster are all chemical. For the CO oxidation by O2 molecules that leads to the formation of C–O bonds and the dissociation of O–O bonds, the Langmuir–Hinshelwood (LH) mechanism is preferred. On the other hand, the Eley–Rideal (ER) mechanism is slightly favored by the oxidation of CO by atomic oxygen. According to the intrinsic reaction coordinate (IRC) calculation, the activation energy for CO oxidation is 4.02 kcal/mol for molecular oxygen and 3.17 kcal/mol for atomic oxygen. Therefore, molecular and atomic oxygen are very reactive for CO oxidation on the Cu19 cluster. To check the applicability of the global hardness response (GHR) profile satisfying the maximum hardness principle along the IRC in the metal cluster reactions, the GHR profile for the oxidation reaction of CO with molecular oxygen and atomic oxygen was computed. The results indicate that this meets the principle of maximum hardness, effectively showcasing the use of DFT methods to analyze the global hardness profile using frontier molecular orbital energy in the context of metal cluster reaction pathways.
Graphical abstract
The CO oxidation by molecular and atomic oxygen on the Cu3 site of the Cu19 cluster employing the density functional theory has been studied. The results show that the CO oxidation by atomic oxygen slightly favors the Eley–Rideal (ER) mechanism, while the CO oxidation by molecular oxygen prefers the Langmuir–Hinshelwood (LH) mechanism. Both molecular and atomic oxygen are very reactive for CO oxidation. Furthermore, the global hardness profile along the intrinsic reaction coordinate follows the maximum hardness principle.
期刊介绍:
Journal of Chemical Sciences is a monthly journal published by the Indian Academy of Sciences. It formed part of the original Proceedings of the Indian Academy of Sciences – Part A, started by the Nobel Laureate Prof C V Raman in 1934, that was split in 1978 into three separate journals. It was renamed as Journal of Chemical Sciences in 2004. The journal publishes original research articles and rapid communications, covering all areas of chemical sciences. A significant feature of the journal is its special issues, brought out from time to time, devoted to conference symposia/proceedings in frontier areas of the subject, held not only in India but also in other countries.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
