{"title":"突尼斯阿尔波特综合征基因研究。","authors":"Mariem El Younsi, Ahlem Achour, Lilia Kraoua, Mezzi Nesrine, Taha Sayari, Ezzeddine Abderrahim, Janet Laabidi, Mohamed Karim Zouaghi, Maher Kharrat, Tahar Gargah, Mediha Trabelsi, Ridha M'rad","doi":"10.1007/s00467-024-06474-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Alport syndrome is a genetic disorder affecting the kidneys, ears, and eyes, causing chronic kidney disease, sensorineural hearing loss, and ocular abnormalities. It results from pathogenic variants in the COL4A3, COL4A4, or COL4A5 genes, with different inheritance patterns: X-linked from COL4A5 variants, autosomal recessive from homozygous variants in COL4A3 or COL4A4, digenic from variants in both COL4A3 and COL4A4, and autosomal dominant from heterozygous variants in COL4A3 or COL4A4.</p><p><strong>Methods: </strong>We analyzed 45 patients with Alport syndrome from 11 Tunisian families to determine their clinical and genetic characteristics. Clinical data were collected retrospectively, and whole-exome sequencing was conducted on one patient from each family. Sanger sequencing validated pathogenic variants, and cascade screening extended the analysis to 53 individuals.</p><p><strong>Results: </strong>We identified nine likely pathogenic variants among 11 index cases: six novel and three known variations. Of these, five were in COL4A3, and four were in COL4A5, with variants including frameshift, nonsense, missense, and alternative splicing. Most variations affected the Gly-XY codon. Among the 45 clinically identified siblings, 30 tested positive for Alport syndrome. The cascade screening identified 3 additional affected individuals, 10 unaffected siblings, and 10 unaffected parents. The mode of inheritance was autosomal recessive in six families and X-linked in four families.</p><p><strong>Conclusions: </strong>This study is the first to screen the mutational spectrum of Alport syndrome in Tunisia. It reveals novel pathogenic variants and suggests that autosomal recessive inheritance may be more common in the Tunisian population than X-linked inheritance, contrary to existing literature.</p>","PeriodicalId":19735,"journal":{"name":"Pediatric Nephrology","volume":" ","pages":"103-116"},"PeriodicalIF":2.5000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Genetic study of Alport syndrome in Tunisia.\",\"authors\":\"Mariem El Younsi, Ahlem Achour, Lilia Kraoua, Mezzi Nesrine, Taha Sayari, Ezzeddine Abderrahim, Janet Laabidi, Mohamed Karim Zouaghi, Maher Kharrat, Tahar Gargah, Mediha Trabelsi, Ridha M'rad\",\"doi\":\"10.1007/s00467-024-06474-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Alport syndrome is a genetic disorder affecting the kidneys, ears, and eyes, causing chronic kidney disease, sensorineural hearing loss, and ocular abnormalities. It results from pathogenic variants in the COL4A3, COL4A4, or COL4A5 genes, with different inheritance patterns: X-linked from COL4A5 variants, autosomal recessive from homozygous variants in COL4A3 or COL4A4, digenic from variants in both COL4A3 and COL4A4, and autosomal dominant from heterozygous variants in COL4A3 or COL4A4.</p><p><strong>Methods: </strong>We analyzed 45 patients with Alport syndrome from 11 Tunisian families to determine their clinical and genetic characteristics. Clinical data were collected retrospectively, and whole-exome sequencing was conducted on one patient from each family. Sanger sequencing validated pathogenic variants, and cascade screening extended the analysis to 53 individuals.</p><p><strong>Results: </strong>We identified nine likely pathogenic variants among 11 index cases: six novel and three known variations. Of these, five were in COL4A3, and four were in COL4A5, with variants including frameshift, nonsense, missense, and alternative splicing. Most variations affected the Gly-XY codon. Among the 45 clinically identified siblings, 30 tested positive for Alport syndrome. The cascade screening identified 3 additional affected individuals, 10 unaffected siblings, and 10 unaffected parents. The mode of inheritance was autosomal recessive in six families and X-linked in four families.</p><p><strong>Conclusions: </strong>This study is the first to screen the mutational spectrum of Alport syndrome in Tunisia. It reveals novel pathogenic variants and suggests that autosomal recessive inheritance may be more common in the Tunisian population than X-linked inheritance, contrary to existing literature.</p>\",\"PeriodicalId\":19735,\"journal\":{\"name\":\"Pediatric Nephrology\",\"volume\":\" \",\"pages\":\"103-116\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pediatric Nephrology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00467-024-06474-7\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/14 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pediatric Nephrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00467-024-06474-7","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/14 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
Background: Alport syndrome is a genetic disorder affecting the kidneys, ears, and eyes, causing chronic kidney disease, sensorineural hearing loss, and ocular abnormalities. It results from pathogenic variants in the COL4A3, COL4A4, or COL4A5 genes, with different inheritance patterns: X-linked from COL4A5 variants, autosomal recessive from homozygous variants in COL4A3 or COL4A4, digenic from variants in both COL4A3 and COL4A4, and autosomal dominant from heterozygous variants in COL4A3 or COL4A4.
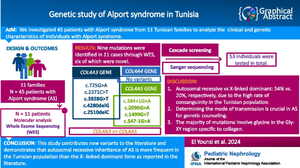
Methods: We analyzed 45 patients with Alport syndrome from 11 Tunisian families to determine their clinical and genetic characteristics. Clinical data were collected retrospectively, and whole-exome sequencing was conducted on one patient from each family. Sanger sequencing validated pathogenic variants, and cascade screening extended the analysis to 53 individuals.
Results: We identified nine likely pathogenic variants among 11 index cases: six novel and three known variations. Of these, five were in COL4A3, and four were in COL4A5, with variants including frameshift, nonsense, missense, and alternative splicing. Most variations affected the Gly-XY codon. Among the 45 clinically identified siblings, 30 tested positive for Alport syndrome. The cascade screening identified 3 additional affected individuals, 10 unaffected siblings, and 10 unaffected parents. The mode of inheritance was autosomal recessive in six families and X-linked in four families.
Conclusions: This study is the first to screen the mutational spectrum of Alport syndrome in Tunisia. It reveals novel pathogenic variants and suggests that autosomal recessive inheritance may be more common in the Tunisian population than X-linked inheritance, contrary to existing literature.
期刊介绍:
International Pediatric Nephrology Association
Pediatric Nephrology publishes original clinical research related to acute and chronic diseases that affect renal function, blood pressure, and fluid and electrolyte disorders in children. Studies may involve medical, surgical, nutritional, physiologic, biochemical, genetic, pathologic or immunologic aspects of disease, imaging techniques or consequences of acute or chronic kidney disease. There are 12 issues per year that contain Editorial Commentaries, Reviews, Educational Reviews, Original Articles, Brief Reports, Rapid Communications, Clinical Quizzes, and Letters to the Editors.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
