{"title":"揭示发育性癫痫脑病 25 型的神经影像学特征:全面回顾已报道病例和一种新型 SLC13A5 变异体。","authors":"Mohammad Farid Mohammadi, Sahand Tehrani Fateh, Maedeh Ganji, Pouria Mohammadi, Tayyeb Bahrami, Mahmoud Reza Ashrafi, Sareh Hosseinpour, Morteza Heidari, Masoud Garshasbi, Ali Reza Tavasoli","doi":"10.1007/s13760-024-02611-z","DOIUrl":null,"url":null,"abstract":"<div><p>Developmental and epileptic encephalopathy type 25 with amelogenesis imperfecta (DEE25) is a rare autosomal recessive disorder caused by homozygous or compound heterozygous disease-causing variants in the <i>SLC13A5</i>. These variants can disrupt energy production and delay brain development, leading to DEE25. Key symptoms include refractory seizures, often manifesting in neonates or infants, alongside global developmental delay, intellectual disability, progressive microcephaly, ataxia, spasticity, and speech difficulties. Dental anomalies related to amelogenesis imperfecta are common. Previous studies have typically reported normal or minimally altered early-life brain magnetic resonance imaging (MRI) findings in DEE25. However, our investigation identified a homozygous splice donor variant (NM_177550.5: c.1437 + 1G >T) in <i>SLC13A5</i> through whole-exome sequencing in two affected siblings (P1 and P2). They displayed developmental delay, cerebral hypotonia, speech delay, recurrent seizures, mild but constant microcephaly, and motor impairments. Significantly, P1 exhibited novel findings on brain magnetic resonance imaging at age 5, including previously unreported extensive persistent hypomyelination. Meanwhile, P2 showed substantial loss of cerebral white matter in the frontoparietal region and delayed myelination at 18 months old. These discoveries broaden the DEE25 imaging spectrum and highlight the clinical heterogeneity even within siblings sharing the same variants.</p></div>","PeriodicalId":7042,"journal":{"name":"Acta neurologica Belgica","volume":"124 6","pages":"1959 - 1972"},"PeriodicalIF":2.0000,"publicationDate":"2024-08-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Unraveling neuroimaging insights in developmental epileptic encephalopathy type 25: a comprehensive review of reported cases and a novel SLC13A5 variant\",\"authors\":\"Mohammad Farid Mohammadi, Sahand Tehrani Fateh, Maedeh Ganji, Pouria Mohammadi, Tayyeb Bahrami, Mahmoud Reza Ashrafi, Sareh Hosseinpour, Morteza Heidari, Masoud Garshasbi, Ali Reza Tavasoli\",\"doi\":\"10.1007/s13760-024-02611-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Developmental and epileptic encephalopathy type 25 with amelogenesis imperfecta (DEE25) is a rare autosomal recessive disorder caused by homozygous or compound heterozygous disease-causing variants in the <i>SLC13A5</i>. These variants can disrupt energy production and delay brain development, leading to DEE25. Key symptoms include refractory seizures, often manifesting in neonates or infants, alongside global developmental delay, intellectual disability, progressive microcephaly, ataxia, spasticity, and speech difficulties. Dental anomalies related to amelogenesis imperfecta are common. Previous studies have typically reported normal or minimally altered early-life brain magnetic resonance imaging (MRI) findings in DEE25. However, our investigation identified a homozygous splice donor variant (NM_177550.5: c.1437 + 1G >T) in <i>SLC13A5</i> through whole-exome sequencing in two affected siblings (P1 and P2). They displayed developmental delay, cerebral hypotonia, speech delay, recurrent seizures, mild but constant microcephaly, and motor impairments. Significantly, P1 exhibited novel findings on brain magnetic resonance imaging at age 5, including previously unreported extensive persistent hypomyelination. Meanwhile, P2 showed substantial loss of cerebral white matter in the frontoparietal region and delayed myelination at 18 months old. These discoveries broaden the DEE25 imaging spectrum and highlight the clinical heterogeneity even within siblings sharing the same variants.</p></div>\",\"PeriodicalId\":7042,\"journal\":{\"name\":\"Acta neurologica Belgica\",\"volume\":\"124 6\",\"pages\":\"1959 - 1972\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-08-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta neurologica Belgica\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s13760-024-02611-z\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta neurologica Belgica","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s13760-024-02611-z","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Unraveling neuroimaging insights in developmental epileptic encephalopathy type 25: a comprehensive review of reported cases and a novel SLC13A5 variant
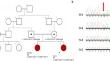
Developmental and epileptic encephalopathy type 25 with amelogenesis imperfecta (DEE25) is a rare autosomal recessive disorder caused by homozygous or compound heterozygous disease-causing variants in the SLC13A5. These variants can disrupt energy production and delay brain development, leading to DEE25. Key symptoms include refractory seizures, often manifesting in neonates or infants, alongside global developmental delay, intellectual disability, progressive microcephaly, ataxia, spasticity, and speech difficulties. Dental anomalies related to amelogenesis imperfecta are common. Previous studies have typically reported normal or minimally altered early-life brain magnetic resonance imaging (MRI) findings in DEE25. However, our investigation identified a homozygous splice donor variant (NM_177550.5: c.1437 + 1G >T) in SLC13A5 through whole-exome sequencing in two affected siblings (P1 and P2). They displayed developmental delay, cerebral hypotonia, speech delay, recurrent seizures, mild but constant microcephaly, and motor impairments. Significantly, P1 exhibited novel findings on brain magnetic resonance imaging at age 5, including previously unreported extensive persistent hypomyelination. Meanwhile, P2 showed substantial loss of cerebral white matter in the frontoparietal region and delayed myelination at 18 months old. These discoveries broaden the DEE25 imaging spectrum and highlight the clinical heterogeneity even within siblings sharing the same variants.
期刊介绍:
Peer-reviewed and published quarterly, Acta Neurologica Belgicapresents original articles in the clinical and basic neurosciences, and also reports the proceedings and the abstracts of the scientific meetings of the different partner societies. The contents include commentaries, editorials, review articles, case reports, neuro-images of interest, book reviews and letters to the editor.
Acta Neurologica Belgica is the official journal of the following national societies:
Belgian Neurological Society
Belgian Society for Neuroscience
Belgian Society of Clinical Neurophysiology
Belgian Pediatric Neurology Society
Belgian Study Group of Multiple Sclerosis
Belgian Stroke Council
Belgian Headache Society
Belgian Study Group of Neuropathology




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
