Kimberly K. O. Walden, Yanghui Cao, Christopher J. Fields, Alvaro G. Hernandez, Gloria A. Rendon, Gene E. Robinson, Rachel K. Skinner, Jeffrey A. Stein, Christopher H. Dietrich
{"title":"代表 6 个目(昆虫纲:鞘翅目、双翅目、半翅目、膜翅目、鳞翅目、神经翅目)的 9 个非模式北美昆虫物种的高质量基因组组装。","authors":"Kimberly K. O. Walden, Yanghui Cao, Christopher J. Fields, Alvaro G. Hernandez, Gloria A. Rendon, Gene E. Robinson, Rachel K. Skinner, Jeffrey A. Stein, Christopher H. Dietrich","doi":"10.1111/1755-0998.14010","DOIUrl":null,"url":null,"abstract":"<p>Field-collected specimens were used to obtain nine high-quality genome assemblies from a total of 10 insect species native to prairies and savannas of central Illinois (USA): <i>Mellilla xanthometata</i> (Lepidoptera: Geometridae), <i>Stenolophus ochropezus</i> (Coleoptera: Carabidae), <i>Forcipata loca</i> (Hemiptera: Cicadellidae), <i>Coelinius</i> sp. (Hymenoptera: Braconidae), <i>Thaumatomyia glabra</i> (Diptera: Chloropidae), <i>Brachynemurus abdominalus</i> (Neuroptera: Myrmeleontidae), <i>Catonia carolina</i> (Hemiptera: Achilidae), <i>Oncometopia orbona</i> (Hemiptera: Cicadellidae), <i>Flexamia atlantica</i> (Hemiptera: Cicadellidae) and <i>Stictocephala bisonia</i> (Hemiptera: Membracidae). Sequencing library preparation from single specimens was successful despite extremely small DNA yields (<0.1 μg) for some samples. Additional sequencing and assembly workflows were adapted to each sample depending on the initial DNA yield. PacBio circular consensus (CCS/HiFi) or continuous long reads (CLR) libraries were used to sequence DNA fragments up to 50 kb in length, with Illumina sequenced linked-reads (TellSeq libraries) and Omni-C libraries used for scaffolding and gap-filling. Assembled genome sizes ranged from 135 MB to 3.2 GB. The number of assembled scaffolds ranged from 47 to >13,000, with the longest scaffold per assembly ranging from ~23 to 439 Mb. Genome completeness was high, with BUSCO scores ranging from 85.5% completeness for the largest genome (<i>Stictocephala bisonia</i>) to 98.8% completeness for the smallest genome (<i>Coelinius</i> sp.). The unique content was estimated using RepeatMasker and GenomeScope2, which ranged from 50.7% to 75.8% and roughly decreased with increasing genome size. Structural annotation predicted a range of 19,281–72,469 protein models for sequenced species. Sequencing costs per genome at the time ranged from US$3–5k, averaged ~1600 CPU-hours on a high-performance cluster and required approximately 14 h of bioinformatics analyses with samples using PacBio HiFi data. Most assemblies would benefit from further manual curation to correct possible scaffold misjoins and translocations suggested by off-diagonal or depleted signals in Omni-C contact maps.</p>","PeriodicalId":211,"journal":{"name":"Molecular Ecology Resources","volume":"24 8","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.14010","citationCount":"0","resultStr":"{\"title\":\"High-quality genome assemblies for nine non-model North American insect species representing six orders (Insecta: Coleoptera, Diptera, Hemiptera, Hymenoptera, Lepidoptera, Neuroptera)\",\"authors\":\"Kimberly K. O. Walden, Yanghui Cao, Christopher J. Fields, Alvaro G. Hernandez, Gloria A. Rendon, Gene E. Robinson, Rachel K. Skinner, Jeffrey A. Stein, Christopher H. Dietrich\",\"doi\":\"10.1111/1755-0998.14010\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Field-collected specimens were used to obtain nine high-quality genome assemblies from a total of 10 insect species native to prairies and savannas of central Illinois (USA): <i>Mellilla xanthometata</i> (Lepidoptera: Geometridae), <i>Stenolophus ochropezus</i> (Coleoptera: Carabidae), <i>Forcipata loca</i> (Hemiptera: Cicadellidae), <i>Coelinius</i> sp. (Hymenoptera: Braconidae), <i>Thaumatomyia glabra</i> (Diptera: Chloropidae), <i>Brachynemurus abdominalus</i> (Neuroptera: Myrmeleontidae), <i>Catonia carolina</i> (Hemiptera: Achilidae), <i>Oncometopia orbona</i> (Hemiptera: Cicadellidae), <i>Flexamia atlantica</i> (Hemiptera: Cicadellidae) and <i>Stictocephala bisonia</i> (Hemiptera: Membracidae). Sequencing library preparation from single specimens was successful despite extremely small DNA yields (<0.1 μg) for some samples. Additional sequencing and assembly workflows were adapted to each sample depending on the initial DNA yield. PacBio circular consensus (CCS/HiFi) or continuous long reads (CLR) libraries were used to sequence DNA fragments up to 50 kb in length, with Illumina sequenced linked-reads (TellSeq libraries) and Omni-C libraries used for scaffolding and gap-filling. Assembled genome sizes ranged from 135 MB to 3.2 GB. The number of assembled scaffolds ranged from 47 to >13,000, with the longest scaffold per assembly ranging from ~23 to 439 Mb. Genome completeness was high, with BUSCO scores ranging from 85.5% completeness for the largest genome (<i>Stictocephala bisonia</i>) to 98.8% completeness for the smallest genome (<i>Coelinius</i> sp.). The unique content was estimated using RepeatMasker and GenomeScope2, which ranged from 50.7% to 75.8% and roughly decreased with increasing genome size. Structural annotation predicted a range of 19,281–72,469 protein models for sequenced species. Sequencing costs per genome at the time ranged from US$3–5k, averaged ~1600 CPU-hours on a high-performance cluster and required approximately 14 h of bioinformatics analyses with samples using PacBio HiFi data. Most assemblies would benefit from further manual curation to correct possible scaffold misjoins and translocations suggested by off-diagonal or depleted signals in Omni-C contact maps.</p>\",\"PeriodicalId\":211,\"journal\":{\"name\":\"Molecular Ecology Resources\",\"volume\":\"24 8\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.14010\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Ecology Resources\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.14010\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Ecology Resources","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.14010","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
High-quality genome assemblies for nine non-model North American insect species representing six orders (Insecta: Coleoptera, Diptera, Hemiptera, Hymenoptera, Lepidoptera, Neuroptera)
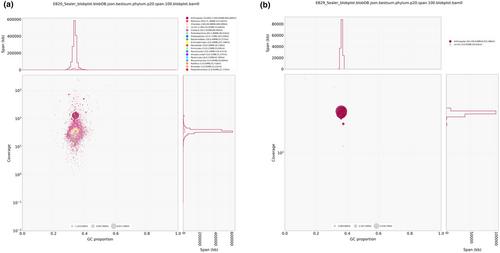
Field-collected specimens were used to obtain nine high-quality genome assemblies from a total of 10 insect species native to prairies and savannas of central Illinois (USA): Mellilla xanthometata (Lepidoptera: Geometridae), Stenolophus ochropezus (Coleoptera: Carabidae), Forcipata loca (Hemiptera: Cicadellidae), Coelinius sp. (Hymenoptera: Braconidae), Thaumatomyia glabra (Diptera: Chloropidae), Brachynemurus abdominalus (Neuroptera: Myrmeleontidae), Catonia carolina (Hemiptera: Achilidae), Oncometopia orbona (Hemiptera: Cicadellidae), Flexamia atlantica (Hemiptera: Cicadellidae) and Stictocephala bisonia (Hemiptera: Membracidae). Sequencing library preparation from single specimens was successful despite extremely small DNA yields (<0.1 μg) for some samples. Additional sequencing and assembly workflows were adapted to each sample depending on the initial DNA yield. PacBio circular consensus (CCS/HiFi) or continuous long reads (CLR) libraries were used to sequence DNA fragments up to 50 kb in length, with Illumina sequenced linked-reads (TellSeq libraries) and Omni-C libraries used for scaffolding and gap-filling. Assembled genome sizes ranged from 135 MB to 3.2 GB. The number of assembled scaffolds ranged from 47 to >13,000, with the longest scaffold per assembly ranging from ~23 to 439 Mb. Genome completeness was high, with BUSCO scores ranging from 85.5% completeness for the largest genome (Stictocephala bisonia) to 98.8% completeness for the smallest genome (Coelinius sp.). The unique content was estimated using RepeatMasker and GenomeScope2, which ranged from 50.7% to 75.8% and roughly decreased with increasing genome size. Structural annotation predicted a range of 19,281–72,469 protein models for sequenced species. Sequencing costs per genome at the time ranged from US$3–5k, averaged ~1600 CPU-hours on a high-performance cluster and required approximately 14 h of bioinformatics analyses with samples using PacBio HiFi data. Most assemblies would benefit from further manual curation to correct possible scaffold misjoins and translocations suggested by off-diagonal or depleted signals in Omni-C contact maps.
期刊介绍:
Molecular Ecology Resources promotes the creation of comprehensive resources for the scientific community, encompassing computer programs, statistical and molecular advancements, and a diverse array of molecular tools. Serving as a conduit for disseminating these resources, the journal targets a broad audience of researchers in the fields of evolution, ecology, and conservation. Articles in Molecular Ecology Resources are crafted to support investigations tackling significant questions within these disciplines.
In addition to original resource articles, Molecular Ecology Resources features Reviews, Opinions, and Comments relevant to the field. The journal also periodically releases Special Issues focusing on resource development within specific areas.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
