{"title":"罕见变异对疾病遗传性的贡献远大于传统估计:等位基因分布模型的修正。","authors":"Yoshiro Nagao","doi":"10.1038/s10038-024-01281-2","DOIUrl":null,"url":null,"abstract":"“Missing heritability” is a current problem in human genetics. I previously reported a method to estimate heritability of a polymorphism (hp2) for a common disease without calculating the genetic variance under dominant and the recessive models. Here, I extend the method to the co-dominant model and carry out trial calculations of hp2. I also calculate hp2 applying the allele distribution model originally reported by Pawitan et al. for comparison as a conventional method. But unexpectedly, hp2 calculated for rare variants with high odds ratios was much higher than the calculated values with the allele distribution model. Also, while examining the basis for the difference in calculated hp2, I noticed that conventional methods use the allele frequency (AF) of a variant in the general population to calculate the genetic variance of that variant. However, this implicitly assumes that the unaffected are included among the phenotypes of the disease – an assumption that is inconsistent with case-control studies in which unaffected individuals belong to the control (unaffected) group. Therefore, I modified the allele distribution model by using the AF in the patient population. Consequently, the hp2 of rare variants calculated with the modified allele distribution model was quite high. Recalculating hp2 of several rare variants reported in the literature with the modified allele distribution model yielded results were 3.2 - 53.7 times higher than the hp2 calculated with the original allele distribution model. These results suggest that the contribution of rare variants to heritability of a disease has been considerably underestimated.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 12","pages":"663-668"},"PeriodicalIF":2.5000,"publicationDate":"2024-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Contribution of rare variants to heritability of a disease is much greater than conventionally estimated: modification of allele distribution model\",\"authors\":\"Yoshiro Nagao\",\"doi\":\"10.1038/s10038-024-01281-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"“Missing heritability” is a current problem in human genetics. I previously reported a method to estimate heritability of a polymorphism (hp2) for a common disease without calculating the genetic variance under dominant and the recessive models. Here, I extend the method to the co-dominant model and carry out trial calculations of hp2. I also calculate hp2 applying the allele distribution model originally reported by Pawitan et al. for comparison as a conventional method. But unexpectedly, hp2 calculated for rare variants with high odds ratios was much higher than the calculated values with the allele distribution model. Also, while examining the basis for the difference in calculated hp2, I noticed that conventional methods use the allele frequency (AF) of a variant in the general population to calculate the genetic variance of that variant. However, this implicitly assumes that the unaffected are included among the phenotypes of the disease – an assumption that is inconsistent with case-control studies in which unaffected individuals belong to the control (unaffected) group. Therefore, I modified the allele distribution model by using the AF in the patient population. Consequently, the hp2 of rare variants calculated with the modified allele distribution model was quite high. Recalculating hp2 of several rare variants reported in the literature with the modified allele distribution model yielded results were 3.2 - 53.7 times higher than the hp2 calculated with the original allele distribution model. These results suggest that the contribution of rare variants to heritability of a disease has been considerably underestimated.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 12\",\"pages\":\"663-668\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-08-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01281-2\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01281-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Contribution of rare variants to heritability of a disease is much greater than conventionally estimated: modification of allele distribution model
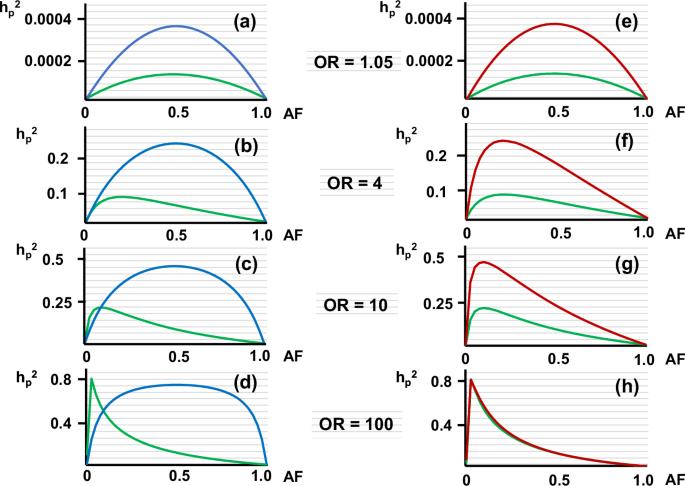
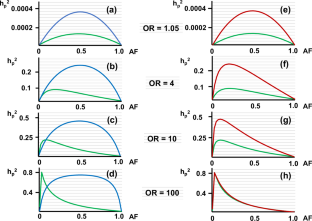
“Missing heritability” is a current problem in human genetics. I previously reported a method to estimate heritability of a polymorphism (hp2) for a common disease without calculating the genetic variance under dominant and the recessive models. Here, I extend the method to the co-dominant model and carry out trial calculations of hp2. I also calculate hp2 applying the allele distribution model originally reported by Pawitan et al. for comparison as a conventional method. But unexpectedly, hp2 calculated for rare variants with high odds ratios was much higher than the calculated values with the allele distribution model. Also, while examining the basis for the difference in calculated hp2, I noticed that conventional methods use the allele frequency (AF) of a variant in the general population to calculate the genetic variance of that variant. However, this implicitly assumes that the unaffected are included among the phenotypes of the disease – an assumption that is inconsistent with case-control studies in which unaffected individuals belong to the control (unaffected) group. Therefore, I modified the allele distribution model by using the AF in the patient population. Consequently, the hp2 of rare variants calculated with the modified allele distribution model was quite high. Recalculating hp2 of several rare variants reported in the literature with the modified allele distribution model yielded results were 3.2 - 53.7 times higher than the hp2 calculated with the original allele distribution model. These results suggest that the contribution of rare variants to heritability of a disease has been considerably underestimated.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
