Jon López-Zorrilla, Xabier M Aretxabaleta, Hegoi Manzano
{"title":"利用迁移学习增强型机器学习原子势探索硅酸二钙的多态性。","authors":"Jon López-Zorrilla, Xabier M Aretxabaleta, Hegoi Manzano","doi":"10.1021/acs.jctc.4c00479","DOIUrl":null,"url":null,"abstract":"<p><p>Belitic cements are a greener alternative to Ordinary Portland Cement due to the lower CO<sub>2</sub> associated with their production. However, their low reactivity with water is currently a drawback, resulting in longer setting times. In this study, we utilize a combination of evolutionary algorithms and machine learning atomic potentials (MLPs) to identify previously unreported belite polymorphs that may exhibit higher hydraulic reactivity than the known phases. To address the high computational demand of this methodology, we propose a novel transfer learning approach for generating MLPs. First, the models are pretrained on a large set of classical data (ReaxFF) and then retrained with density functional theory (DFT) data. We demonstrate that the transfer learning enhanced potentials exhibit higher accuracy, require less training data, and are more transferable than those trained exclusively on DFT data. The generated machine learning potential enables a fast, exhaustive, and reliable exploration of the dicalcium silicate polymorphs. This includes studying their stability through phonon analysis and calculating their structural and elastic properties. Overall, we identify ten new belite polymorphs within the energy range of the existing ones, including a layered phase with potentially high reactivity.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"7682-7690"},"PeriodicalIF":5.5000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring the Polymorphism of Dicalcium Silicates Using Transfer Learning Enhanced Machine Learning Atomic Potentials.\",\"authors\":\"Jon López-Zorrilla, Xabier M Aretxabaleta, Hegoi Manzano\",\"doi\":\"10.1021/acs.jctc.4c00479\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Belitic cements are a greener alternative to Ordinary Portland Cement due to the lower CO<sub>2</sub> associated with their production. However, their low reactivity with water is currently a drawback, resulting in longer setting times. In this study, we utilize a combination of evolutionary algorithms and machine learning atomic potentials (MLPs) to identify previously unreported belite polymorphs that may exhibit higher hydraulic reactivity than the known phases. To address the high computational demand of this methodology, we propose a novel transfer learning approach for generating MLPs. First, the models are pretrained on a large set of classical data (ReaxFF) and then retrained with density functional theory (DFT) data. We demonstrate that the transfer learning enhanced potentials exhibit higher accuracy, require less training data, and are more transferable than those trained exclusively on DFT data. The generated machine learning potential enables a fast, exhaustive, and reliable exploration of the dicalcium silicate polymorphs. This includes studying their stability through phonon analysis and calculating their structural and elastic properties. Overall, we identify ten new belite polymorphs within the energy range of the existing ones, including a layered phase with potentially high reactivity.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"7682-7690\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00479\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00479","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/22 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Exploring the Polymorphism of Dicalcium Silicates Using Transfer Learning Enhanced Machine Learning Atomic Potentials.
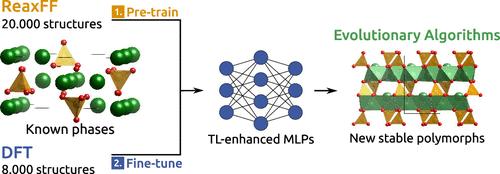
Belitic cements are a greener alternative to Ordinary Portland Cement due to the lower CO2 associated with their production. However, their low reactivity with water is currently a drawback, resulting in longer setting times. In this study, we utilize a combination of evolutionary algorithms and machine learning atomic potentials (MLPs) to identify previously unreported belite polymorphs that may exhibit higher hydraulic reactivity than the known phases. To address the high computational demand of this methodology, we propose a novel transfer learning approach for generating MLPs. First, the models are pretrained on a large set of classical data (ReaxFF) and then retrained with density functional theory (DFT) data. We demonstrate that the transfer learning enhanced potentials exhibit higher accuracy, require less training data, and are more transferable than those trained exclusively on DFT data. The generated machine learning potential enables a fast, exhaustive, and reliable exploration of the dicalcium silicate polymorphs. This includes studying their stability through phonon analysis and calculating their structural and elastic properties. Overall, we identify ten new belite polymorphs within the energy range of the existing ones, including a layered phase with potentially high reactivity.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
