Mohamed Ibrahim , Xinyuanyuan Sun , Vinicius Martins de Oliveira , Ruibin Liu , Joseph Clayton , Haifa El Kilani , Jana Shen , Rolf Hilgenfeld
{"title":"为什么 SARS-CoV-2 的 Omicron 主蛋白酶不如野生型蛋白酶稳定?晶体学、生物物理和理论研究","authors":"Mohamed Ibrahim , Xinyuanyuan Sun , Vinicius Martins de Oliveira , Ruibin Liu , Joseph Clayton , Haifa El Kilani , Jana Shen , Rolf Hilgenfeld","doi":"10.1016/j.hlife.2024.06.003","DOIUrl":null,"url":null,"abstract":"<div><p>During the continuing evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)<strong>,</strong> the Omicron variant of concern emerged in the second half of 2021 and has been dominant since November of that year. Along with its sublineages, it has maintained a prominent role ever since. The Nsp5 main protease (M<sup>pro</sup>) of the Omicron virus is characterized by a single dominant mutation, P132H. Here we determined the X-ray crystal structures of the P132H mutant (or O-M<sup>pro</sup>) as a free enzyme and in complex with the M<sup>pro</sup> inhibitor, the alpha-ketoamide 13b-K, and we conducted enzymological, biophysical, as well as theoretical studies to characterize the O-M<sup>pro</sup>. We found that O-M<sup>pro</sup> has a similar overall structure and binding with 13b-K; however, it displays lower enzymatic activity and lower thermal stability compared to the WT-M<sup>pro</sup> (with “WT” referring to the prototype strain). Intriguingly, the imidazole ring of His132 and the carboxylate plane of Glu240 are in a stacked configuration in the X-ray structures determined here. Empirical folding free energy calculations suggest that the O-M<sup>pro</sup> dimer is destabilized relative to the WT-M<sup>pro</sup> due to less favorable van der Waals interactions and backbone conformations in the individual protomers. All-atom continuous constant-pH molecular dynamics (MD) simulations reveal that His132 and Glu240 display coupled titration. At pH 7, His132 is predominantly neutral and in a stacked configuration with respect to Glu240 which is charged. In order to examine whether the Omicron mutation eases the emergence of further M<sup>pro</sup> mutations, we also analyzed the P132H+T169S double mutant, which is characteristic of the BA.1.1.2 lineage. However, we found little evidence of a correlation between the two mutation sites.</p></div>","PeriodicalId":100609,"journal":{"name":"hLife","volume":"2 8","pages":"Pages 419-433"},"PeriodicalIF":0.0000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S294992832400049X/pdfft?md5=46f4a86fab2e946cbb89c2ad34803747&pid=1-s2.0-S294992832400049X-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Why is the Omicron main protease of SARS-CoV-2 less stable than its wild-type counterpart? A crystallographic, biophysical, and theoretical study\",\"authors\":\"Mohamed Ibrahim , Xinyuanyuan Sun , Vinicius Martins de Oliveira , Ruibin Liu , Joseph Clayton , Haifa El Kilani , Jana Shen , Rolf Hilgenfeld\",\"doi\":\"10.1016/j.hlife.2024.06.003\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>During the continuing evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)<strong>,</strong> the Omicron variant of concern emerged in the second half of 2021 and has been dominant since November of that year. Along with its sublineages, it has maintained a prominent role ever since. The Nsp5 main protease (M<sup>pro</sup>) of the Omicron virus is characterized by a single dominant mutation, P132H. Here we determined the X-ray crystal structures of the P132H mutant (or O-M<sup>pro</sup>) as a free enzyme and in complex with the M<sup>pro</sup> inhibitor, the alpha-ketoamide 13b-K, and we conducted enzymological, biophysical, as well as theoretical studies to characterize the O-M<sup>pro</sup>. We found that O-M<sup>pro</sup> has a similar overall structure and binding with 13b-K; however, it displays lower enzymatic activity and lower thermal stability compared to the WT-M<sup>pro</sup> (with “WT” referring to the prototype strain). Intriguingly, the imidazole ring of His132 and the carboxylate plane of Glu240 are in a stacked configuration in the X-ray structures determined here. Empirical folding free energy calculations suggest that the O-M<sup>pro</sup> dimer is destabilized relative to the WT-M<sup>pro</sup> due to less favorable van der Waals interactions and backbone conformations in the individual protomers. All-atom continuous constant-pH molecular dynamics (MD) simulations reveal that His132 and Glu240 display coupled titration. At pH 7, His132 is predominantly neutral and in a stacked configuration with respect to Glu240 which is charged. In order to examine whether the Omicron mutation eases the emergence of further M<sup>pro</sup> mutations, we also analyzed the P132H+T169S double mutant, which is characteristic of the BA.1.1.2 lineage. However, we found little evidence of a correlation between the two mutation sites.</p></div>\",\"PeriodicalId\":100609,\"journal\":{\"name\":\"hLife\",\"volume\":\"2 8\",\"pages\":\"Pages 419-433\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S294992832400049X/pdfft?md5=46f4a86fab2e946cbb89c2ad34803747&pid=1-s2.0-S294992832400049X-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"hLife\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S294992832400049X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"hLife","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S294992832400049X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/15 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Why is the Omicron main protease of SARS-CoV-2 less stable than its wild-type counterpart? A crystallographic, biophysical, and theoretical study
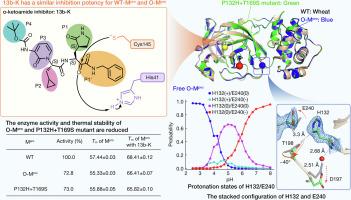
During the continuing evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the Omicron variant of concern emerged in the second half of 2021 and has been dominant since November of that year. Along with its sublineages, it has maintained a prominent role ever since. The Nsp5 main protease (Mpro) of the Omicron virus is characterized by a single dominant mutation, P132H. Here we determined the X-ray crystal structures of the P132H mutant (or O-Mpro) as a free enzyme and in complex with the Mpro inhibitor, the alpha-ketoamide 13b-K, and we conducted enzymological, biophysical, as well as theoretical studies to characterize the O-Mpro. We found that O-Mpro has a similar overall structure and binding with 13b-K; however, it displays lower enzymatic activity and lower thermal stability compared to the WT-Mpro (with “WT” referring to the prototype strain). Intriguingly, the imidazole ring of His132 and the carboxylate plane of Glu240 are in a stacked configuration in the X-ray structures determined here. Empirical folding free energy calculations suggest that the O-Mpro dimer is destabilized relative to the WT-Mpro due to less favorable van der Waals interactions and backbone conformations in the individual protomers. All-atom continuous constant-pH molecular dynamics (MD) simulations reveal that His132 and Glu240 display coupled titration. At pH 7, His132 is predominantly neutral and in a stacked configuration with respect to Glu240 which is charged. In order to examine whether the Omicron mutation eases the emergence of further Mpro mutations, we also analyzed the P132H+T169S double mutant, which is characteristic of the BA.1.1.2 lineage. However, we found little evidence of a correlation between the two mutation sites.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
