{"title":"通过分离分析发现的导致非综合征性胸主动脉瘤和夹层的新缺失MYLK变异体","authors":"Daigo Nishijo, Hiroki Yagi, Nana Akiyama, Norifumi Takeda, Masahiko Ando, Haruo Yamauchi, Norihiko Takeda, Issei Komuro","doi":"10.1155/2024/4281972","DOIUrl":null,"url":null,"abstract":"<p><p>Nonsyndromic hereditary thoracic aortic aneurysm and dissection (TAAD) is an autosomal dominant disease; however, it is frequently difficult to identify the causative genes. We report in this study a 33-year-old Japanese male with TAAD (Stanford type A) that is complicated with severe aortic regurgitation. There was no family history of aortic diseases in the patient nor any specific clinical features suggestive of connective tissue diseases, such as Marfan syndrome. Genetic testing identified candidate causative variants in two different genes: <i>MYLK</i> (c.4819G > A, p.[Gly1607Ser]) and <i>FBN1</i> (c.365G > A, p.[Arg122His]). Familial cosegregation analysis revealed that the novel de novo <i>MYLK</i> variant was present only in the proband, and the <i>FBN1</i> variant was also found in his nonaffected mother, and thus the <i>MYLK</i> variant was classified as likely pathogenic. <i>MYLK</i> is a causative gene for nonsyndromic TAAD that requires careful management; however, the number of reports is limited. Accumulating data on the pathogenicity of rare variants by performing a comprehensive pedigree analysis would help establish better treatment strategies for life-threatening hereditary TAAD cases.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2024 ","pages":"4281972"},"PeriodicalIF":0.0000,"publicationDate":"2024-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11343627/pdf/","citationCount":"0","resultStr":"{\"title\":\"A <i>De Novo</i> Missense <i>MYLK</i> Variant Leading to Nonsyndromic Thoracic Aortic Aneurysm and Dissection Identified by Segregation Analysis.\",\"authors\":\"Daigo Nishijo, Hiroki Yagi, Nana Akiyama, Norifumi Takeda, Masahiko Ando, Haruo Yamauchi, Norihiko Takeda, Issei Komuro\",\"doi\":\"10.1155/2024/4281972\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Nonsyndromic hereditary thoracic aortic aneurysm and dissection (TAAD) is an autosomal dominant disease; however, it is frequently difficult to identify the causative genes. We report in this study a 33-year-old Japanese male with TAAD (Stanford type A) that is complicated with severe aortic regurgitation. There was no family history of aortic diseases in the patient nor any specific clinical features suggestive of connective tissue diseases, such as Marfan syndrome. Genetic testing identified candidate causative variants in two different genes: <i>MYLK</i> (c.4819G > A, p.[Gly1607Ser]) and <i>FBN1</i> (c.365G > A, p.[Arg122His]). Familial cosegregation analysis revealed that the novel de novo <i>MYLK</i> variant was present only in the proband, and the <i>FBN1</i> variant was also found in his nonaffected mother, and thus the <i>MYLK</i> variant was classified as likely pathogenic. <i>MYLK</i> is a causative gene for nonsyndromic TAAD that requires careful management; however, the number of reports is limited. Accumulating data on the pathogenicity of rare variants by performing a comprehensive pedigree analysis would help establish better treatment strategies for life-threatening hereditary TAAD cases.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\"2024 \",\"pages\":\"4281972\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11343627/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2024/4281972\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2024/4281972","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
A De Novo Missense MYLK Variant Leading to Nonsyndromic Thoracic Aortic Aneurysm and Dissection Identified by Segregation Analysis.
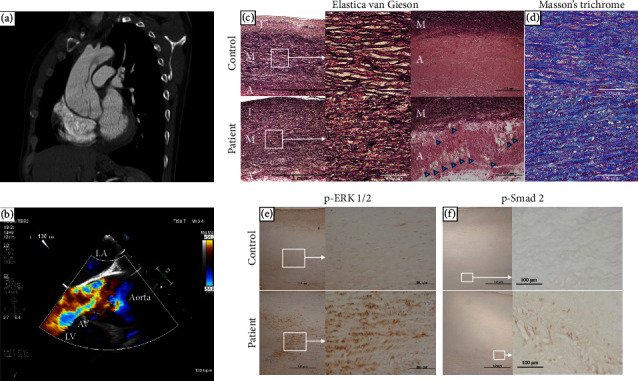
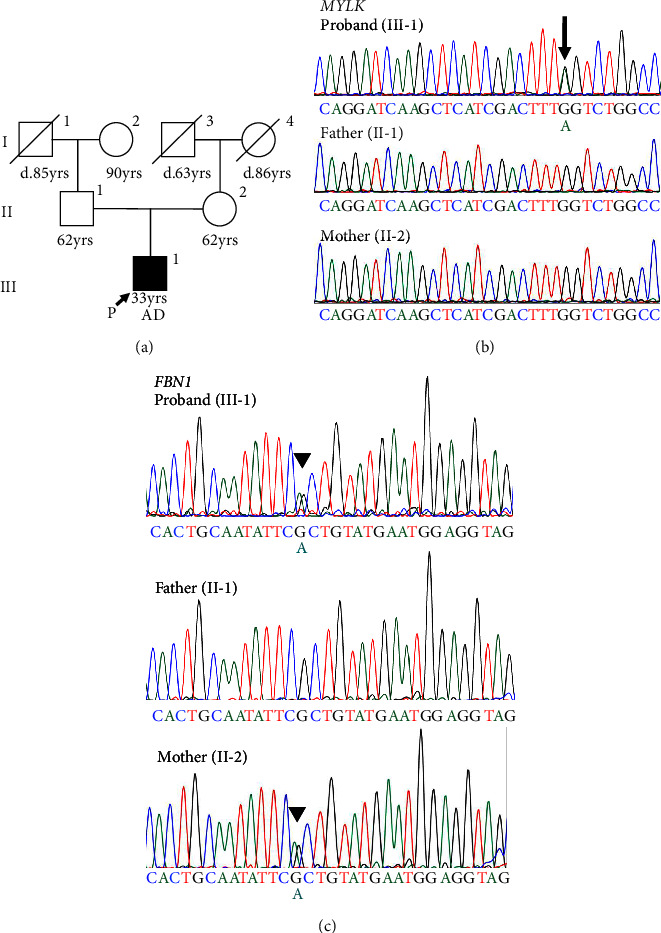
Nonsyndromic hereditary thoracic aortic aneurysm and dissection (TAAD) is an autosomal dominant disease; however, it is frequently difficult to identify the causative genes. We report in this study a 33-year-old Japanese male with TAAD (Stanford type A) that is complicated with severe aortic regurgitation. There was no family history of aortic diseases in the patient nor any specific clinical features suggestive of connective tissue diseases, such as Marfan syndrome. Genetic testing identified candidate causative variants in two different genes: MYLK (c.4819G > A, p.[Gly1607Ser]) and FBN1 (c.365G > A, p.[Arg122His]). Familial cosegregation analysis revealed that the novel de novo MYLK variant was present only in the proband, and the FBN1 variant was also found in his nonaffected mother, and thus the MYLK variant was classified as likely pathogenic. MYLK is a causative gene for nonsyndromic TAAD that requires careful management; however, the number of reports is limited. Accumulating data on the pathogenicity of rare variants by performing a comprehensive pedigree analysis would help establish better treatment strategies for life-threatening hereditary TAAD cases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
