{"title":"光解模型:甲醇的量子动力学模拟。","authors":"Léon L E Cigrang, Graham A Worth","doi":"10.1021/acs.jpca.4c03612","DOIUrl":null,"url":null,"abstract":"<p><p>A comprehensive computational study of the gas-phase photodissociation dynamics of methanol is presented. Using a multiconfigurational active space based method (RASSCF) to obtain multidimensional potential energy surfaces (PESs) on-the-fly, direct quantum dynamics simulations were run using the variational multi-configurational Gaussian method (DD-vMCG). Different initial excitation energies were simulated to investigate the dependence of the branching ratios on the electronic state being populated. A detailed mechanistic explanation is provided for the observed differences with respect to the excitation energy. Population of the lowest lying excited state of methanol leads to rapid hydroxyl hydrogen loss as the main dissociation channel. This is rationalized by the strongly dissociative nature of the PES cut along the O-H stretching coordinate, confirmed by the broad feature in the absorption spectrum. In contrast, more energetic excitations lead mainly to C-O bond breaking. Again, analysis of the diabatic surfaces offers a clear explanation in terms of the nature of the electronic states involved and the coupling between them. The type of calculations presented, as well as the subsequent analysis of the results, should be seen as a general workflow for the modeling of photochemical reactions.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":" ","pages":"7546-7557"},"PeriodicalIF":2.8000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11403662/pdf/","citationCount":"0","resultStr":"{\"title\":\"Modeling Photodissociation: Quantum Dynamics Simulations of Methanol.\",\"authors\":\"Léon L E Cigrang, Graham A Worth\",\"doi\":\"10.1021/acs.jpca.4c03612\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A comprehensive computational study of the gas-phase photodissociation dynamics of methanol is presented. Using a multiconfigurational active space based method (RASSCF) to obtain multidimensional potential energy surfaces (PESs) on-the-fly, direct quantum dynamics simulations were run using the variational multi-configurational Gaussian method (DD-vMCG). Different initial excitation energies were simulated to investigate the dependence of the branching ratios on the electronic state being populated. A detailed mechanistic explanation is provided for the observed differences with respect to the excitation energy. Population of the lowest lying excited state of methanol leads to rapid hydroxyl hydrogen loss as the main dissociation channel. This is rationalized by the strongly dissociative nature of the PES cut along the O-H stretching coordinate, confirmed by the broad feature in the absorption spectrum. In contrast, more energetic excitations lead mainly to C-O bond breaking. Again, analysis of the diabatic surfaces offers a clear explanation in terms of the nature of the electronic states involved and the coupling between them. The type of calculations presented, as well as the subsequent analysis of the results, should be seen as a general workflow for the modeling of photochemical reactions.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\" \",\"pages\":\"7546-7557\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11403662/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpca.4c03612\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c03612","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
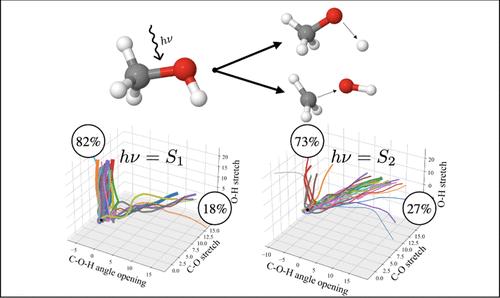
本文对甲醇的气相光解动力学进行了全面的计算研究。利用基于多配置活动空间的方法(RASSCF)即时获得多维势能面(PES),并使用变异多配置高斯方法(DD-vMCG)进行直接量子动力学模拟。模拟了不同的初始激发能量,以研究分支率对填充的电子状态的依赖性。对观察到的激发能量差异提供了详细的机理解释。甲醇最低激发态的填充导致羟基氢迅速丧失,成为主要的解离通道。这可以从 PES 沿着 O-H 伸展坐标切割的强解离性质得到合理解释,吸收光谱中的宽特征也证实了这一点。相比之下,能量更高的激发主要导致 C-O 键断裂。同样,对二重态表面的分析也从所涉及的电子态性质以及它们之间的耦合角度提供了清晰的解释。所介绍的计算类型以及随后对结果的分析应被视为光化学反应建模的一般工作流程。
Modeling Photodissociation: Quantum Dynamics Simulations of Methanol.
A comprehensive computational study of the gas-phase photodissociation dynamics of methanol is presented. Using a multiconfigurational active space based method (RASSCF) to obtain multidimensional potential energy surfaces (PESs) on-the-fly, direct quantum dynamics simulations were run using the variational multi-configurational Gaussian method (DD-vMCG). Different initial excitation energies were simulated to investigate the dependence of the branching ratios on the electronic state being populated. A detailed mechanistic explanation is provided for the observed differences with respect to the excitation energy. Population of the lowest lying excited state of methanol leads to rapid hydroxyl hydrogen loss as the main dissociation channel. This is rationalized by the strongly dissociative nature of the PES cut along the O-H stretching coordinate, confirmed by the broad feature in the absorption spectrum. In contrast, more energetic excitations lead mainly to C-O bond breaking. Again, analysis of the diabatic surfaces offers a clear explanation in terms of the nature of the electronic states involved and the coupling between them. The type of calculations presented, as well as the subsequent analysis of the results, should be seen as a general workflow for the modeling of photochemical reactions.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
