Yu-Jen Lin, Arul S Menon, Zhiqiang Hu, Steven E Brenner
{"title":"变异影响预测数据库(VIPdb),第 2 版:三十年来遗传变异影响预测的趋势。","authors":"Yu-Jen Lin, Arul S Menon, Zhiqiang Hu, Steven E Brenner","doi":"10.1186/s40246-024-00663-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Variant interpretation is essential for identifying patients' disease-causing genetic variants amongst the millions detected in their genomes. Hundreds of Variant Impact Predictors (VIPs), also known as Variant Effect Predictors (VEPs), have been developed for this purpose, with a variety of methodologies and goals. To facilitate the exploration of available VIP options, we have created the Variant Impact Predictor database (VIPdb).</p><p><strong>Results: </strong>The Variant Impact Predictor database (VIPdb) version 2 presents a collection of VIPs developed over the past three decades, summarizing their characteristics, ClinGen calibrated scores, CAGI assessment results, publication details, access information, and citation patterns. We previously summarized 217 VIPs and their features in VIPdb in 2019. Building upon this foundation, we identified and categorized an additional 190 VIPs, resulting in a total of 407 VIPs in VIPdb version 2. The majority of the VIPs have the capacity to predict the impacts of single nucleotide variants and nonsynonymous variants. More VIPs tailored to predict the impacts of insertions and deletions have been developed since the 2010s. In contrast, relatively few VIPs are dedicated to the prediction of splicing, structural, synonymous, and regulatory variants. The increasing rate of citations to VIPs reflects the ongoing growth in their use, and the evolving trends in citations reveal development in the field and individual methods.</p><p><strong>Conclusions: </strong>VIPdb version 2 summarizes 407 VIPs and their features, potentially facilitating VIP exploration for various variant interpretation applications. VIPdb is available at https://genomeinterpretation.org/vipdb.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"18 1","pages":"90"},"PeriodicalIF":4.3000,"publicationDate":"2024-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11360829/pdf/","citationCount":"0","resultStr":"{\"title\":\"Variant Impact Predictor database (VIPdb), version 2: trends from three decades of genetic variant impact predictors.\",\"authors\":\"Yu-Jen Lin, Arul S Menon, Zhiqiang Hu, Steven E Brenner\",\"doi\":\"10.1186/s40246-024-00663-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Variant interpretation is essential for identifying patients' disease-causing genetic variants amongst the millions detected in their genomes. Hundreds of Variant Impact Predictors (VIPs), also known as Variant Effect Predictors (VEPs), have been developed for this purpose, with a variety of methodologies and goals. To facilitate the exploration of available VIP options, we have created the Variant Impact Predictor database (VIPdb).</p><p><strong>Results: </strong>The Variant Impact Predictor database (VIPdb) version 2 presents a collection of VIPs developed over the past three decades, summarizing their characteristics, ClinGen calibrated scores, CAGI assessment results, publication details, access information, and citation patterns. We previously summarized 217 VIPs and their features in VIPdb in 2019. Building upon this foundation, we identified and categorized an additional 190 VIPs, resulting in a total of 407 VIPs in VIPdb version 2. The majority of the VIPs have the capacity to predict the impacts of single nucleotide variants and nonsynonymous variants. More VIPs tailored to predict the impacts of insertions and deletions have been developed since the 2010s. In contrast, relatively few VIPs are dedicated to the prediction of splicing, structural, synonymous, and regulatory variants. The increasing rate of citations to VIPs reflects the ongoing growth in their use, and the evolving trends in citations reveal development in the field and individual methods.</p><p><strong>Conclusions: </strong>VIPdb version 2 summarizes 407 VIPs and their features, potentially facilitating VIP exploration for various variant interpretation applications. VIPdb is available at https://genomeinterpretation.org/vipdb.</p>\",\"PeriodicalId\":13183,\"journal\":{\"name\":\"Human Genomics\",\"volume\":\"18 1\",\"pages\":\"90\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11360829/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s40246-024-00663-z\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-024-00663-z","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Variant Impact Predictor database (VIPdb), version 2: trends from three decades of genetic variant impact predictors.
Background: Variant interpretation is essential for identifying patients' disease-causing genetic variants amongst the millions detected in their genomes. Hundreds of Variant Impact Predictors (VIPs), also known as Variant Effect Predictors (VEPs), have been developed for this purpose, with a variety of methodologies and goals. To facilitate the exploration of available VIP options, we have created the Variant Impact Predictor database (VIPdb).
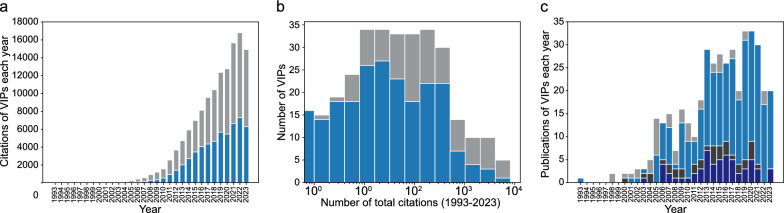
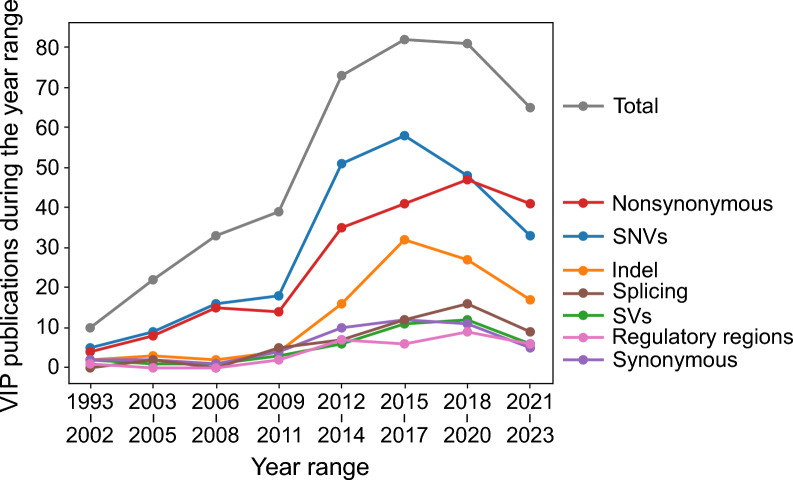
Results: The Variant Impact Predictor database (VIPdb) version 2 presents a collection of VIPs developed over the past three decades, summarizing their characteristics, ClinGen calibrated scores, CAGI assessment results, publication details, access information, and citation patterns. We previously summarized 217 VIPs and their features in VIPdb in 2019. Building upon this foundation, we identified and categorized an additional 190 VIPs, resulting in a total of 407 VIPs in VIPdb version 2. The majority of the VIPs have the capacity to predict the impacts of single nucleotide variants and nonsynonymous variants. More VIPs tailored to predict the impacts of insertions and deletions have been developed since the 2010s. In contrast, relatively few VIPs are dedicated to the prediction of splicing, structural, synonymous, and regulatory variants. The increasing rate of citations to VIPs reflects the ongoing growth in their use, and the evolving trends in citations reveal development in the field and individual methods.
Conclusions: VIPdb version 2 summarizes 407 VIPs and their features, potentially facilitating VIP exploration for various variant interpretation applications. VIPdb is available at https://genomeinterpretation.org/vipdb.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
