Jeanne Jury, Jean-François Benoist, Madeleine Joubert, Chloé Quelin, Thomas Besnard, Solène Conrad, Claudine Le Vaillant, Stéphane Bézieau, Bertrand Isidor, Tania Attié-Bitach, Benjamin Cogné, Marie Vincent
{"title":"两个谷胱甘肽合成酶缺乏症(GSS)胎儿的多种先天性畸形。","authors":"Jeanne Jury, Jean-François Benoist, Madeleine Joubert, Chloé Quelin, Thomas Besnard, Solène Conrad, Claudine Le Vaillant, Stéphane Bézieau, Bertrand Isidor, Tania Attié-Bitach, Benjamin Cogné, Marie Vincent","doi":"10.1111/cge.14613","DOIUrl":null,"url":null,"abstract":"<p>Glutathione synthetase deficiency is a rare inborn metabolic disease usually caused by biallelic variants in <i>GSS</i>. Clinical severity varies from isolated hemolytic anemia, sometimes associated with chronic metabolic acidosis and 5-oxoprolinuria, to severe neurological phenotypes with neonatal lethality. Here we report on two fetal siblings from two pregnancies with glutathione synthetase deficiency exhibiting similar multiple congenital anomalies associating phocomelia, cleft palate, intra-uterine growth retardation, genito-urinary malformations, and congenital heart defect. Genome sequencing showed that both fetuses were compound heterozygous for two <i>GSS</i> variants: the previously reported pathogenic missense substitution NM_000178.4 c.800G>A p.(Arg267Gln), and a 2.4 kb intragenic deletion NC_000020.11:g.34944530_34946833del. RNA-seq on brain tissue revealed the out-of-frame deletion of the exon 3 and an almost monoallelic expression of the missense variant (88%), suggesting degradation of the deletion-harboring allele by nonsense-mediated mRNA decay. 5-oxoproline (pyroglutamic acid) levels in amniotic fluid were elevated, suggesting an alteration of the gamma-glutamyl cycle, and corroborating the pathogenicity of the two <i>GSS</i> variants. Only one case of glutathione synthetase deficiency with limb malformations has previously been reported, in a newborn homozygous for the c.800G>A variant. Thus, our data allow us to discuss a potential phenotypic extension of glutathione synthetase deficiency, with a possible involvement of the c.800G>A variant.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 6","pages":"776-781"},"PeriodicalIF":2.3000,"publicationDate":"2024-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14613","citationCount":"0","resultStr":"{\"title\":\"Multiple congenital anomalies in two fetuses with glutathione-synthetase deficit (GSS)\",\"authors\":\"Jeanne Jury, Jean-François Benoist, Madeleine Joubert, Chloé Quelin, Thomas Besnard, Solène Conrad, Claudine Le Vaillant, Stéphane Bézieau, Bertrand Isidor, Tania Attié-Bitach, Benjamin Cogné, Marie Vincent\",\"doi\":\"10.1111/cge.14613\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Glutathione synthetase deficiency is a rare inborn metabolic disease usually caused by biallelic variants in <i>GSS</i>. Clinical severity varies from isolated hemolytic anemia, sometimes associated with chronic metabolic acidosis and 5-oxoprolinuria, to severe neurological phenotypes with neonatal lethality. Here we report on two fetal siblings from two pregnancies with glutathione synthetase deficiency exhibiting similar multiple congenital anomalies associating phocomelia, cleft palate, intra-uterine growth retardation, genito-urinary malformations, and congenital heart defect. Genome sequencing showed that both fetuses were compound heterozygous for two <i>GSS</i> variants: the previously reported pathogenic missense substitution NM_000178.4 c.800G>A p.(Arg267Gln), and a 2.4 kb intragenic deletion NC_000020.11:g.34944530_34946833del. RNA-seq on brain tissue revealed the out-of-frame deletion of the exon 3 and an almost monoallelic expression of the missense variant (88%), suggesting degradation of the deletion-harboring allele by nonsense-mediated mRNA decay. 5-oxoproline (pyroglutamic acid) levels in amniotic fluid were elevated, suggesting an alteration of the gamma-glutamyl cycle, and corroborating the pathogenicity of the two <i>GSS</i> variants. Only one case of glutathione synthetase deficiency with limb malformations has previously been reported, in a newborn homozygous for the c.800G>A variant. Thus, our data allow us to discuss a potential phenotypic extension of glutathione synthetase deficiency, with a possible involvement of the c.800G>A variant.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":\"106 6\",\"pages\":\"776-781\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14613\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14613\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14613","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Multiple congenital anomalies in two fetuses with glutathione-synthetase deficit (GSS)
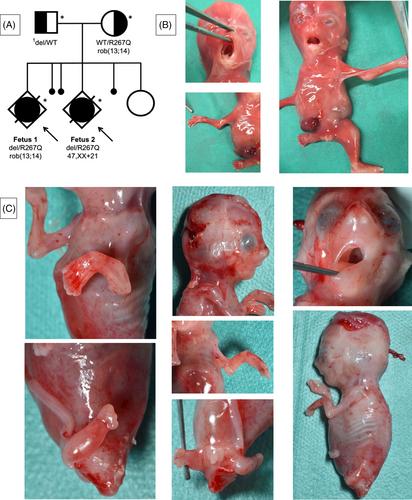
Glutathione synthetase deficiency is a rare inborn metabolic disease usually caused by biallelic variants in GSS. Clinical severity varies from isolated hemolytic anemia, sometimes associated with chronic metabolic acidosis and 5-oxoprolinuria, to severe neurological phenotypes with neonatal lethality. Here we report on two fetal siblings from two pregnancies with glutathione synthetase deficiency exhibiting similar multiple congenital anomalies associating phocomelia, cleft palate, intra-uterine growth retardation, genito-urinary malformations, and congenital heart defect. Genome sequencing showed that both fetuses were compound heterozygous for two GSS variants: the previously reported pathogenic missense substitution NM_000178.4 c.800G>A p.(Arg267Gln), and a 2.4 kb intragenic deletion NC_000020.11:g.34944530_34946833del. RNA-seq on brain tissue revealed the out-of-frame deletion of the exon 3 and an almost monoallelic expression of the missense variant (88%), suggesting degradation of the deletion-harboring allele by nonsense-mediated mRNA decay. 5-oxoproline (pyroglutamic acid) levels in amniotic fluid were elevated, suggesting an alteration of the gamma-glutamyl cycle, and corroborating the pathogenicity of the two GSS variants. Only one case of glutathione synthetase deficiency with limb malformations has previously been reported, in a newborn homozygous for the c.800G>A variant. Thus, our data allow us to discuss a potential phenotypic extension of glutathione synthetase deficiency, with a possible involvement of the c.800G>A variant.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
