Rida Fatima , A. Afaq , Muhammad Ahmed , Abdul Quader , Abu Bakar , Abdulmohsen Alruwaili
{"title":"用多种 DFT 方法探究 AgYF3(Y=镁、锶)卤化物包晶的结构和光电特性的第一性原理见解","authors":"Rida Fatima , A. Afaq , Muhammad Ahmed , Abdul Quader , Abu Bakar , Abdulmohsen Alruwaili","doi":"10.1016/j.chemphys.2024.112443","DOIUrl":null,"url":null,"abstract":"<div><p>This study reports the structural, electronic, optical, phonon, thermodynamic and thermoelectric properties of <span><math><msub><mrow><mi>AgYF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> (X=Mg, Sr) for photovoltaic and energy applications. We performed first principles calculations using full potential linearized augmented plane wave, FP-LAPW method implemented in Wien2k. The generalized gradient approximations of Perdew–Burke–Ernzerhof PBE-GGA, and PBE revised for solids, PBEsol, is employed for structural optimization of these lead free halide perovskites. The Birch–Murnaghan energy volume curve fitting comprehend the structural stability. The optimized lattice constant of <span><math><msub><mrow><mi>AgMgF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> and <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> obtained with PBE-GGA(PBEsol) is 3.99(3.92)<!--> <!-->Å and 4.42(4.65)<!--> <!-->Å. The stability is further tested with the help of formation energy and positive phonon dispersion curves calculations. For the calculations of explicit electronic and optical properties, we also employed Tran–Blaha modified Beck–Johnson (TB-mBJ) and Strongly Constrained but Appropriately Normed, SCAN, exchange and correlations functionals. The electronic band gap of <span><math><msub><mrow><mi>AgMgF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> computed with PBEsol, TB-mBJ and SCAN is 1.96 eV, 5.25 eV and 2.59 eV exhibiting M-<span><math><mi>Γ</mi></math></span> indirect band gap. The band gap energy of <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> is 2.06 eV, 6.42 eV and 2.70 eV with PBEsol, TB-mBJ and SCAN. The indirect band gap nature of <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> is confirmed by PBEsol and TB-mBJ while it anticipated direct band gap behavior with meta-GGA SCAN. The different optical parameters like dielectric constant, optical conductivity, energy loss function, absorption, reflectivity and refractive index are calculated to assess optical activity of both perovskites. Comprehensive electronic and optical analysis advocates the utility of <span><math><msub><mrow><mi>AgMgF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> and <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> for different applications is solar technology and optoelectronic devices.</p></div>","PeriodicalId":272,"journal":{"name":"Chemical Physics","volume":"588 ","pages":"Article 112443"},"PeriodicalIF":2.8000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"First-Principles insights to probe structural and opto-electronic properties of AgYF3 (Y=Mg, Sr) halide perovskites with variety of DFT methods\",\"authors\":\"Rida Fatima , A. Afaq , Muhammad Ahmed , Abdul Quader , Abu Bakar , Abdulmohsen Alruwaili\",\"doi\":\"10.1016/j.chemphys.2024.112443\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>This study reports the structural, electronic, optical, phonon, thermodynamic and thermoelectric properties of <span><math><msub><mrow><mi>AgYF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> (X=Mg, Sr) for photovoltaic and energy applications. We performed first principles calculations using full potential linearized augmented plane wave, FP-LAPW method implemented in Wien2k. The generalized gradient approximations of Perdew–Burke–Ernzerhof PBE-GGA, and PBE revised for solids, PBEsol, is employed for structural optimization of these lead free halide perovskites. The Birch–Murnaghan energy volume curve fitting comprehend the structural stability. The optimized lattice constant of <span><math><msub><mrow><mi>AgMgF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> and <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> obtained with PBE-GGA(PBEsol) is 3.99(3.92)<!--> <!-->Å and 4.42(4.65)<!--> <!-->Å. The stability is further tested with the help of formation energy and positive phonon dispersion curves calculations. For the calculations of explicit electronic and optical properties, we also employed Tran–Blaha modified Beck–Johnson (TB-mBJ) and Strongly Constrained but Appropriately Normed, SCAN, exchange and correlations functionals. The electronic band gap of <span><math><msub><mrow><mi>AgMgF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> computed with PBEsol, TB-mBJ and SCAN is 1.96 eV, 5.25 eV and 2.59 eV exhibiting M-<span><math><mi>Γ</mi></math></span> indirect band gap. The band gap energy of <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> is 2.06 eV, 6.42 eV and 2.70 eV with PBEsol, TB-mBJ and SCAN. The indirect band gap nature of <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> is confirmed by PBEsol and TB-mBJ while it anticipated direct band gap behavior with meta-GGA SCAN. The different optical parameters like dielectric constant, optical conductivity, energy loss function, absorption, reflectivity and refractive index are calculated to assess optical activity of both perovskites. Comprehensive electronic and optical analysis advocates the utility of <span><math><msub><mrow><mi>AgMgF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> and <span><math><msub><mrow><mi>AgSrF</mi></mrow><mrow><mn>3</mn></mrow></msub></math></span> for different applications is solar technology and optoelectronic devices.</p></div>\",\"PeriodicalId\":272,\"journal\":{\"name\":\"Chemical Physics\",\"volume\":\"588 \",\"pages\":\"Article 112443\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0301010424002726\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301010424002726","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/30 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
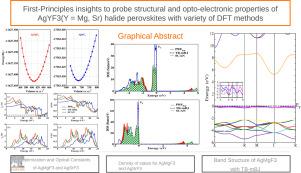
First-Principles insights to probe structural and opto-electronic properties of AgYF3 (Y=Mg, Sr) halide perovskites with variety of DFT methods
This study reports the structural, electronic, optical, phonon, thermodynamic and thermoelectric properties of (X=Mg, Sr) for photovoltaic and energy applications. We performed first principles calculations using full potential linearized augmented plane wave, FP-LAPW method implemented in Wien2k. The generalized gradient approximations of Perdew–Burke–Ernzerhof PBE-GGA, and PBE revised for solids, PBEsol, is employed for structural optimization of these lead free halide perovskites. The Birch–Murnaghan energy volume curve fitting comprehend the structural stability. The optimized lattice constant of and obtained with PBE-GGA(PBEsol) is 3.99(3.92) Å and 4.42(4.65) Å. The stability is further tested with the help of formation energy and positive phonon dispersion curves calculations. For the calculations of explicit electronic and optical properties, we also employed Tran–Blaha modified Beck–Johnson (TB-mBJ) and Strongly Constrained but Appropriately Normed, SCAN, exchange and correlations functionals. The electronic band gap of computed with PBEsol, TB-mBJ and SCAN is 1.96 eV, 5.25 eV and 2.59 eV exhibiting M- indirect band gap. The band gap energy of is 2.06 eV, 6.42 eV and 2.70 eV with PBEsol, TB-mBJ and SCAN. The indirect band gap nature of is confirmed by PBEsol and TB-mBJ while it anticipated direct band gap behavior with meta-GGA SCAN. The different optical parameters like dielectric constant, optical conductivity, energy loss function, absorption, reflectivity and refractive index are calculated to assess optical activity of both perovskites. Comprehensive electronic and optical analysis advocates the utility of and for different applications is solar technology and optoelectronic devices.
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
