Shengchun Wang, Xu Luo, Yuan Wang, Zhao Liu, Yi Yu, Xuejie Wang, Demin Ren, Pengjie Wang, Yi-Hung Chen, Xiaotian Qi, Hong Yi, Aiwen Lei
{"title":"由自由基引发的 C-C 双键和官能团易位","authors":"Shengchun Wang, Xu Luo, Yuan Wang, Zhao Liu, Yi Yu, Xuejie Wang, Demin Ren, Pengjie Wang, Yi-Hung Chen, Xiaotian Qi, Hong Yi, Aiwen Lei","doi":"10.1038/s41557-024-01633-7","DOIUrl":null,"url":null,"abstract":"Multi-site functionalization of molecules provides a potent approach to accessing intricate compounds. However, simultaneous functionalization of the reactive site and the inert remote C(sp3)–H poses a formidable challenge, as chemical reactions conventionally occur at the most active site. In addition, achieving precise control over site selectivity for remote C(sp3)–H activation presents an additional hurdle. Here we report an alternative modular method for alkene difunctionalization, encompassing radical-triggered translocation of functional groups and remote C(sp3)–H desaturation via photo/cobalt dual catalysis. By systematically combining radical addition, functional group migration and cobalt-promoted hydrogen atom transfer, we successfully effectuate the translocation of the carbon–carbon double bond and another functional group with precise site selectivity and remarkable E/Z selectivity. This redox-neutral approach shows good compatibility with diverse fluoroalkyl and sulfonyl radical precursors, enabling the migration of benzoyloxy, acetoxy, formyl, cyano and heteroaryl groups. This protocol offers a resolution for the simultaneous transformation of manifold sites. Simultaneous functionalization of reactive and inert remote sites presents a powerful approach to access complex molecules, yet it is hindered by precise remote C(sp3)–H activation. Now, the accurate translocation of functional groups and remote C–H desaturation has been achieved in parallel through combining functional group migration and cobalt-promoted hydrogen atom transfer.","PeriodicalId":18909,"journal":{"name":"Nature chemistry","volume":"16 10","pages":"1621-1629"},"PeriodicalIF":20.2000,"publicationDate":"2024-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Radical-triggered translocation of C–C double bond and functional group\",\"authors\":\"Shengchun Wang, Xu Luo, Yuan Wang, Zhao Liu, Yi Yu, Xuejie Wang, Demin Ren, Pengjie Wang, Yi-Hung Chen, Xiaotian Qi, Hong Yi, Aiwen Lei\",\"doi\":\"10.1038/s41557-024-01633-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Multi-site functionalization of molecules provides a potent approach to accessing intricate compounds. However, simultaneous functionalization of the reactive site and the inert remote C(sp3)–H poses a formidable challenge, as chemical reactions conventionally occur at the most active site. In addition, achieving precise control over site selectivity for remote C(sp3)–H activation presents an additional hurdle. Here we report an alternative modular method for alkene difunctionalization, encompassing radical-triggered translocation of functional groups and remote C(sp3)–H desaturation via photo/cobalt dual catalysis. By systematically combining radical addition, functional group migration and cobalt-promoted hydrogen atom transfer, we successfully effectuate the translocation of the carbon–carbon double bond and another functional group with precise site selectivity and remarkable E/Z selectivity. This redox-neutral approach shows good compatibility with diverse fluoroalkyl and sulfonyl radical precursors, enabling the migration of benzoyloxy, acetoxy, formyl, cyano and heteroaryl groups. This protocol offers a resolution for the simultaneous transformation of manifold sites. Simultaneous functionalization of reactive and inert remote sites presents a powerful approach to access complex molecules, yet it is hindered by precise remote C(sp3)–H activation. Now, the accurate translocation of functional groups and remote C–H desaturation has been achieved in parallel through combining functional group migration and cobalt-promoted hydrogen atom transfer.\",\"PeriodicalId\":18909,\"journal\":{\"name\":\"Nature chemistry\",\"volume\":\"16 10\",\"pages\":\"1621-1629\"},\"PeriodicalIF\":20.2000,\"publicationDate\":\"2024-09-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.nature.com/articles/s41557-024-01633-7\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.nature.com/articles/s41557-024-01633-7","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
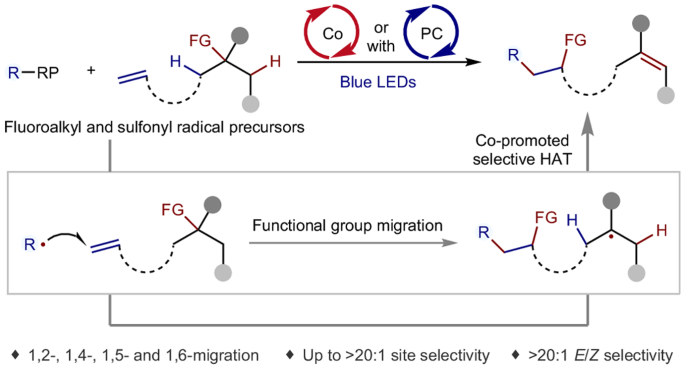
Radical-triggered translocation of C–C double bond and functional group
Multi-site functionalization of molecules provides a potent approach to accessing intricate compounds. However, simultaneous functionalization of the reactive site and the inert remote C(sp3)–H poses a formidable challenge, as chemical reactions conventionally occur at the most active site. In addition, achieving precise control over site selectivity for remote C(sp3)–H activation presents an additional hurdle. Here we report an alternative modular method for alkene difunctionalization, encompassing radical-triggered translocation of functional groups and remote C(sp3)–H desaturation via photo/cobalt dual catalysis. By systematically combining radical addition, functional group migration and cobalt-promoted hydrogen atom transfer, we successfully effectuate the translocation of the carbon–carbon double bond and another functional group with precise site selectivity and remarkable E/Z selectivity. This redox-neutral approach shows good compatibility with diverse fluoroalkyl and sulfonyl radical precursors, enabling the migration of benzoyloxy, acetoxy, formyl, cyano and heteroaryl groups. This protocol offers a resolution for the simultaneous transformation of manifold sites. Simultaneous functionalization of reactive and inert remote sites presents a powerful approach to access complex molecules, yet it is hindered by precise remote C(sp3)–H activation. Now, the accurate translocation of functional groups and remote C–H desaturation has been achieved in parallel through combining functional group migration and cobalt-promoted hydrogen atom transfer.
期刊介绍:
Nature Chemistry is a monthly journal that publishes groundbreaking and significant research in all areas of chemistry. It covers traditional subjects such as analytical, inorganic, organic, and physical chemistry, as well as a wide range of other topics including catalysis, computational and theoretical chemistry, and environmental chemistry.
The journal also features interdisciplinary research at the interface of chemistry with biology, materials science, nanotechnology, and physics. Manuscripts detailing such multidisciplinary work are encouraged, as long as the central theme pertains to chemistry.
Aside from primary research, Nature Chemistry publishes review articles, news and views, research highlights from other journals, commentaries, book reviews, correspondence, and analysis of the broader chemical landscape. It also addresses crucial issues related to education, funding, policy, intellectual property, and the societal impact of chemistry.
Nature Chemistry is dedicated to ensuring the highest standards of original research through a fair and rigorous review process. It offers authors maximum visibility for their papers, access to a broad readership, exceptional copy editing and production standards, rapid publication, and independence from academic societies and other vested interests.
Overall, Nature Chemistry aims to be the authoritative voice of the global chemical community.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
