{"title":"使用广义活动空间驱动相似性重正化群的核电离能基准研究","authors":"Meng Huang, Francesco A. Evangelista","doi":"10.1021/acs.jctc.4c00835","DOIUrl":null,"url":null,"abstract":"X-ray photoelectron spectroscopy (XPS) is a powerful experimental technique for probing the electronic structure of molecules and materials; however, interpreting XPS data requires accurate computational methods to model core-ionized states. This work proposes and benchmarks a new approach based on the generalized active space-driven similarity renormalization group (GAS-DSRG) for calculating core-ionization energies and treating correlation effects at the perturbative and nonperturbative levels. We tested the GAS-DSRG across three data sets. First, the vertical core-ionization energies of small molecules containing first-row elements are evaluated. GAS-DSRG achieves mean absolute errors below 0.3 eV, which is comparable to high-level coupled cluster methods. Next, the accuracy of GAS-DSRG is evaluated for larger organic molecules using the CORE65 data set, with the DSRG-MRPT3 level yielding a mean absolute error of only 0.34 eV for 65 core-ionization transitions. Insights are provided into the treatment of static and dynamic correlation, the importance of high-order perturbation theory, and notable differences from density functional theory in the predicted energy ordering of core-ionized states for specific molecules. Finally, vibrationally resolved XPS spectra of diatomic molecules (CO, N<sub>2</sub>, and O<sub>2</sub>) are simulated, showing excellent agreement with experimental data.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"69 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Benchmark Study of Core-Ionization Energies with the Generalized Active Space-Driven Similarity Renormalization Group\",\"authors\":\"Meng Huang, Francesco A. Evangelista\",\"doi\":\"10.1021/acs.jctc.4c00835\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"X-ray photoelectron spectroscopy (XPS) is a powerful experimental technique for probing the electronic structure of molecules and materials; however, interpreting XPS data requires accurate computational methods to model core-ionized states. This work proposes and benchmarks a new approach based on the generalized active space-driven similarity renormalization group (GAS-DSRG) for calculating core-ionization energies and treating correlation effects at the perturbative and nonperturbative levels. We tested the GAS-DSRG across three data sets. First, the vertical core-ionization energies of small molecules containing first-row elements are evaluated. GAS-DSRG achieves mean absolute errors below 0.3 eV, which is comparable to high-level coupled cluster methods. Next, the accuracy of GAS-DSRG is evaluated for larger organic molecules using the CORE65 data set, with the DSRG-MRPT3 level yielding a mean absolute error of only 0.34 eV for 65 core-ionization transitions. Insights are provided into the treatment of static and dynamic correlation, the importance of high-order perturbation theory, and notable differences from density functional theory in the predicted energy ordering of core-ionized states for specific molecules. Finally, vibrationally resolved XPS spectra of diatomic molecules (CO, N<sub>2</sub>, and O<sub>2</sub>) are simulated, showing excellent agreement with experimental data.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"69 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00835\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00835","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Benchmark Study of Core-Ionization Energies with the Generalized Active Space-Driven Similarity Renormalization Group
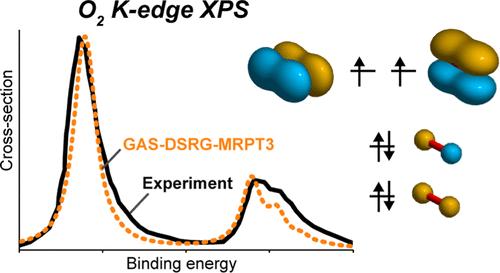
X-ray photoelectron spectroscopy (XPS) is a powerful experimental technique for probing the electronic structure of molecules and materials; however, interpreting XPS data requires accurate computational methods to model core-ionized states. This work proposes and benchmarks a new approach based on the generalized active space-driven similarity renormalization group (GAS-DSRG) for calculating core-ionization energies and treating correlation effects at the perturbative and nonperturbative levels. We tested the GAS-DSRG across three data sets. First, the vertical core-ionization energies of small molecules containing first-row elements are evaluated. GAS-DSRG achieves mean absolute errors below 0.3 eV, which is comparable to high-level coupled cluster methods. Next, the accuracy of GAS-DSRG is evaluated for larger organic molecules using the CORE65 data set, with the DSRG-MRPT3 level yielding a mean absolute error of only 0.34 eV for 65 core-ionization transitions. Insights are provided into the treatment of static and dynamic correlation, the importance of high-order perturbation theory, and notable differences from density functional theory in the predicted energy ordering of core-ionized states for specific molecules. Finally, vibrationally resolved XPS spectra of diatomic molecules (CO, N2, and O2) are simulated, showing excellent agreement with experimental data.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
