Danai Katsanevaki, Sally M. Till, Ingrid Buller-Peralta, Mohammad Sarfaraz Nawaz, Susana R. Louros, Vijayakumar Kapgal, Shashank Tiwari, Darren Walsh, Natasha J. Anstey, Nina G. Petrović, Alison Cormack, Vanesa Salazar-Sanchez, Anjanette Harris, William Farnworth-Rowson, Andrew Sutherland, Thomas C. Watson, Siyan Dimitrov, Adam D. Jackson, Daisy Arkell, Suryanarayan Biswal, Peter C. Kind
{"title":"C2/GAP 结构域在 SYNGAP1 相关病理生理学中的关键作用","authors":"Danai Katsanevaki, Sally M. Till, Ingrid Buller-Peralta, Mohammad Sarfaraz Nawaz, Susana R. Louros, Vijayakumar Kapgal, Shashank Tiwari, Darren Walsh, Natasha J. Anstey, Nina G. Petrović, Alison Cormack, Vanesa Salazar-Sanchez, Anjanette Harris, William Farnworth-Rowson, Andrew Sutherland, Thomas C. Watson, Siyan Dimitrov, Adam D. Jackson, Daisy Arkell, Suryanarayan Biswal, Peter C. Kind","doi":"10.1016/j.celrep.2024.114733","DOIUrl":null,"url":null,"abstract":"<p>Mutations in <em>SYNGAP1</em> are a common genetic cause of intellectual disability (ID) and a risk factor for autism. <em>SYNGAP1</em> encodes a synaptic GTPase-activating protein (GAP) that has both signaling and scaffolding roles. Most pathogenic variants of <em>SYNGAP1</em> are predicted to result in haploinsufficiency. However, some affected individuals carry missense mutations in its calcium/lipid binding (C2) and GAP domains, suggesting that many clinical features result from loss of functions carried out by these domains. To test this hypothesis, we targeted the exons encoding the C2 and GAP domains of SYNGAP. Rats heterozygous for this deletion exhibit reduced exploration and fear extinction, altered social investigation, and spontaneous seizures—key phenotypes shared with <em>Syngap</em> heterozygous null rats. Together, these findings indicate that the reduction of SYNGAP C2/GAP domain function is a main feature of SYNGAP haploinsufficiency. This rat model provides an important system for the study of ID, autism, and epilepsy.</p>","PeriodicalId":9798,"journal":{"name":"Cell reports","volume":"40 1","pages":""},"PeriodicalIF":6.9000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Key roles of C2/GAP domains in SYNGAP1-related pathophysiology\",\"authors\":\"Danai Katsanevaki, Sally M. Till, Ingrid Buller-Peralta, Mohammad Sarfaraz Nawaz, Susana R. Louros, Vijayakumar Kapgal, Shashank Tiwari, Darren Walsh, Natasha J. Anstey, Nina G. Petrović, Alison Cormack, Vanesa Salazar-Sanchez, Anjanette Harris, William Farnworth-Rowson, Andrew Sutherland, Thomas C. Watson, Siyan Dimitrov, Adam D. Jackson, Daisy Arkell, Suryanarayan Biswal, Peter C. Kind\",\"doi\":\"10.1016/j.celrep.2024.114733\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Mutations in <em>SYNGAP1</em> are a common genetic cause of intellectual disability (ID) and a risk factor for autism. <em>SYNGAP1</em> encodes a synaptic GTPase-activating protein (GAP) that has both signaling and scaffolding roles. Most pathogenic variants of <em>SYNGAP1</em> are predicted to result in haploinsufficiency. However, some affected individuals carry missense mutations in its calcium/lipid binding (C2) and GAP domains, suggesting that many clinical features result from loss of functions carried out by these domains. To test this hypothesis, we targeted the exons encoding the C2 and GAP domains of SYNGAP. Rats heterozygous for this deletion exhibit reduced exploration and fear extinction, altered social investigation, and spontaneous seizures—key phenotypes shared with <em>Syngap</em> heterozygous null rats. Together, these findings indicate that the reduction of SYNGAP C2/GAP domain function is a main feature of SYNGAP haploinsufficiency. This rat model provides an important system for the study of ID, autism, and epilepsy.</p>\",\"PeriodicalId\":9798,\"journal\":{\"name\":\"Cell reports\",\"volume\":\"40 1\",\"pages\":\"\"},\"PeriodicalIF\":6.9000,\"publicationDate\":\"2024-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cell reports\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1016/j.celrep.2024.114733\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell reports","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.celrep.2024.114733","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
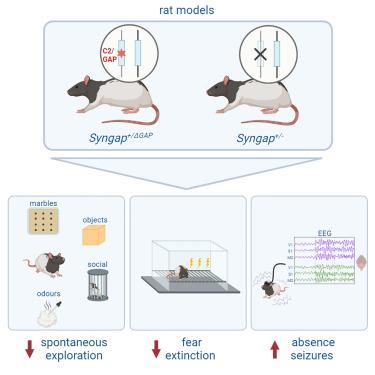
SYNGAP1 基因突变是导致智力障碍(ID)的常见遗传原因,也是自闭症的风险因素之一。SYNGAP1编码一种突触GTP酶激活蛋白(GAP),具有信号传导和支架作用。据预测,SYNGAP1 的大多数致病变体都会导致单倍功能缺陷。然而,一些受影响的个体在其钙/脂结合(C2)和 GAP 结构域中携带错义突变,这表明许多临床特征是由这些结构域的功能缺失造成的。为了验证这一假设,我们以编码 SYNGAP 的 C2 和 GAP 结构域的外显子为靶点。杂合子缺失的大鼠表现出探索和恐惧消退减少、社会调查改变和自发性癫痫发作--这些都是与SYNGAP杂合子无效大鼠相同的主要表型。这些发现共同表明,SYNGAP C2/GAP结构域功能的降低是SYNGAP单倍体缺陷的主要特征。这种大鼠模型为研究ID、自闭症和癫痫提供了一个重要的系统。
Key roles of C2/GAP domains in SYNGAP1-related pathophysiology
Mutations in SYNGAP1 are a common genetic cause of intellectual disability (ID) and a risk factor for autism. SYNGAP1 encodes a synaptic GTPase-activating protein (GAP) that has both signaling and scaffolding roles. Most pathogenic variants of SYNGAP1 are predicted to result in haploinsufficiency. However, some affected individuals carry missense mutations in its calcium/lipid binding (C2) and GAP domains, suggesting that many clinical features result from loss of functions carried out by these domains. To test this hypothesis, we targeted the exons encoding the C2 and GAP domains of SYNGAP. Rats heterozygous for this deletion exhibit reduced exploration and fear extinction, altered social investigation, and spontaneous seizures—key phenotypes shared with Syngap heterozygous null rats. Together, these findings indicate that the reduction of SYNGAP C2/GAP domain function is a main feature of SYNGAP haploinsufficiency. This rat model provides an important system for the study of ID, autism, and epilepsy.
期刊介绍:
Cell Reports publishes high-quality research across the life sciences and focuses on new biological insight as its primary criterion for publication. The journal offers three primary article types: Reports, which are shorter single-point articles, research articles, which are longer and provide deeper mechanistic insights, and resources, which highlight significant technical advances or major informational datasets that contribute to biological advances. Reviews covering recent literature in emerging and active fields are also accepted.
The Cell Reports Portfolio includes gold open-access journals that cover life, medical, and physical sciences, and its mission is to make cutting-edge research and methodologies available to a wide readership.
The journal's professional in-house editors work closely with authors, reviewers, and the scientific advisory board, which consists of current and future leaders in their respective fields. The advisory board guides the scope, content, and quality of the journal, but editorial decisions are independently made by the in-house scientific editors of Cell Reports.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
