Widad Abou Chaar MD, Anirudh N. Eranki, Hannah A. Stevens, Sonya L. Watson MS, Darice Y. Wong PhD, Veronica S. Avila, Megan Delfeld, Alexander J. Gary, Sanjukta Tawde MS, Malia Triebold MS, Marcello Cherchi MD, PhD, Tao Xie MD, PhD, Paul J. Lockhart PhD, Melanie Bahlo PhD, David Pellerin MD, MSc, Marie-Josée Dicaire, Matt Danzi PhD, Stephan Zuchner MD, PhD, Bernard C. Brais MD, PhD, Susan Perlman MD, Margit Burmeister PhD, Henry Paulson MD, PhD, Sharan Srinivasan MD, PhD, Lawrence Schut MD, Matthew Bower MS, Khalaf Bushara MD, Chuanhong Liao MS, Vikram G. Shakkottai MD, PhD, John Collins MD, PhD, H. Brent Clark MD, PhD, Soma Das PhD, Brent L. Fogel MD, PhD, Christopher M. Gomez MD, PhD
{"title":"美国脊髓小脑共济失调(SCA27B)大样本的临床、放射学和病理学特征","authors":"Widad Abou Chaar MD, Anirudh N. Eranki, Hannah A. Stevens, Sonya L. Watson MS, Darice Y. Wong PhD, Veronica S. Avila, Megan Delfeld, Alexander J. Gary, Sanjukta Tawde MS, Malia Triebold MS, Marcello Cherchi MD, PhD, Tao Xie MD, PhD, Paul J. Lockhart PhD, Melanie Bahlo PhD, David Pellerin MD, MSc, Marie-Josée Dicaire, Matt Danzi PhD, Stephan Zuchner MD, PhD, Bernard C. Brais MD, PhD, Susan Perlman MD, Margit Burmeister PhD, Henry Paulson MD, PhD, Sharan Srinivasan MD, PhD, Lawrence Schut MD, Matthew Bower MS, Khalaf Bushara MD, Chuanhong Liao MS, Vikram G. Shakkottai MD, PhD, John Collins MD, PhD, H. Brent Clark MD, PhD, Soma Das PhD, Brent L. Fogel MD, PhD, Christopher M. Gomez MD, PhD","doi":"10.1002/ana.27060","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n <h3> Objectives</h3>\n \n <p>Spinocerebellar ataxia 27B due to GAA repeat expansions in the fibroblast growth factor 14 (<i>FGF14</i>) gene has recently been recognized as a common cause of late-onset hereditary cerebellar ataxia. Here we present the first report of this disease in the US population, characterizing its clinical manifestations, disease progression, pathological abnormalities, and response to 4-aminopyridine in a cohort of 102 patients bearing GAA repeat expansions.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>We compiled a series of patients with SCA27B, recruited from 5 academic centers across the United States. Clinical manifestations and patient demographics were collected retrospectively from clinical records in an unblinded approach using a standardized form. Post-mortem analysis was done on 4 brains of patients with genetically confirmed SCA27B.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>In our cohort of 102 patients with SCA27B, we found that SCA27B was a late-onset (57 ± 12.5 years) slowly progressive ataxia with an episodic component in 51% of patients. Balance and gait impairment were almost always present at disease onset. The principal finding on post-mortem examination of 4 brain specimens was loss of Purkinje neurons that was most severe in the vermis most particularly in the anterior vermis. Similar to European populations, a high percent of patients 21/28 (75%) reported a positive treatment response with 4-aminopyridine.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>Our study further estimates prevalence and further expands the clinical, imaging and pathological features of SCA27B, while looking at treatment response, disease progression, and survival in patients with this disease. Testing for SCA27B should be considered in all undiagnosed ataxia patients, especially those with episodic onset. ANN NEUROL 2024;96:1092–1103</p>\n </section>\n </div>","PeriodicalId":127,"journal":{"name":"Annals of Neurology","volume":"96 6","pages":"1092-1103"},"PeriodicalIF":7.7000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ana.27060","citationCount":"0","resultStr":"{\"title\":\"Clinical, Radiological and Pathological Features of a Large American Cohort of Spinocerebellar Ataxia (SCA27B)\",\"authors\":\"Widad Abou Chaar MD, Anirudh N. Eranki, Hannah A. Stevens, Sonya L. Watson MS, Darice Y. Wong PhD, Veronica S. Avila, Megan Delfeld, Alexander J. Gary, Sanjukta Tawde MS, Malia Triebold MS, Marcello Cherchi MD, PhD, Tao Xie MD, PhD, Paul J. Lockhart PhD, Melanie Bahlo PhD, David Pellerin MD, MSc, Marie-Josée Dicaire, Matt Danzi PhD, Stephan Zuchner MD, PhD, Bernard C. Brais MD, PhD, Susan Perlman MD, Margit Burmeister PhD, Henry Paulson MD, PhD, Sharan Srinivasan MD, PhD, Lawrence Schut MD, Matthew Bower MS, Khalaf Bushara MD, Chuanhong Liao MS, Vikram G. Shakkottai MD, PhD, John Collins MD, PhD, H. Brent Clark MD, PhD, Soma Das PhD, Brent L. Fogel MD, PhD, Christopher M. Gomez MD, PhD\",\"doi\":\"10.1002/ana.27060\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <section>\\n \\n <h3> Objectives</h3>\\n \\n <p>Spinocerebellar ataxia 27B due to GAA repeat expansions in the fibroblast growth factor 14 (<i>FGF14</i>) gene has recently been recognized as a common cause of late-onset hereditary cerebellar ataxia. Here we present the first report of this disease in the US population, characterizing its clinical manifestations, disease progression, pathological abnormalities, and response to 4-aminopyridine in a cohort of 102 patients bearing GAA repeat expansions.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>We compiled a series of patients with SCA27B, recruited from 5 academic centers across the United States. Clinical manifestations and patient demographics were collected retrospectively from clinical records in an unblinded approach using a standardized form. Post-mortem analysis was done on 4 brains of patients with genetically confirmed SCA27B.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>In our cohort of 102 patients with SCA27B, we found that SCA27B was a late-onset (57 ± 12.5 years) slowly progressive ataxia with an episodic component in 51% of patients. Balance and gait impairment were almost always present at disease onset. The principal finding on post-mortem examination of 4 brain specimens was loss of Purkinje neurons that was most severe in the vermis most particularly in the anterior vermis. Similar to European populations, a high percent of patients 21/28 (75%) reported a positive treatment response with 4-aminopyridine.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Interpretation</h3>\\n \\n <p>Our study further estimates prevalence and further expands the clinical, imaging and pathological features of SCA27B, while looking at treatment response, disease progression, and survival in patients with this disease. Testing for SCA27B should be considered in all undiagnosed ataxia patients, especially those with episodic onset. ANN NEUROL 2024;96:1092–1103</p>\\n </section>\\n </div>\",\"PeriodicalId\":127,\"journal\":{\"name\":\"Annals of Neurology\",\"volume\":\"96 6\",\"pages\":\"1092-1103\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2024-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ana.27060\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ana.27060\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Neurology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ana.27060","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Clinical, Radiological and Pathological Features of a Large American Cohort of Spinocerebellar Ataxia (SCA27B)
Objectives
Spinocerebellar ataxia 27B due to GAA repeat expansions in the fibroblast growth factor 14 (FGF14) gene has recently been recognized as a common cause of late-onset hereditary cerebellar ataxia. Here we present the first report of this disease in the US population, characterizing its clinical manifestations, disease progression, pathological abnormalities, and response to 4-aminopyridine in a cohort of 102 patients bearing GAA repeat expansions.
Methods
We compiled a series of patients with SCA27B, recruited from 5 academic centers across the United States. Clinical manifestations and patient demographics were collected retrospectively from clinical records in an unblinded approach using a standardized form. Post-mortem analysis was done on 4 brains of patients with genetically confirmed SCA27B.
Results
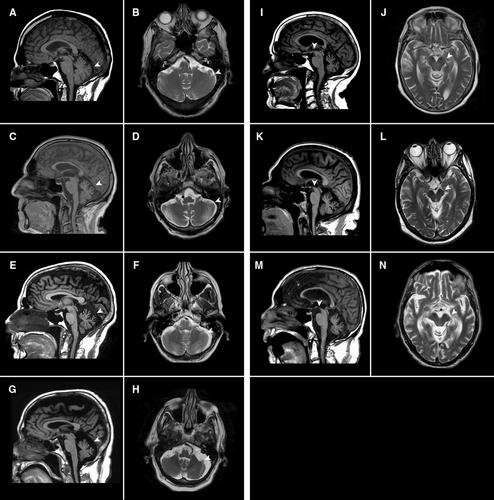
In our cohort of 102 patients with SCA27B, we found that SCA27B was a late-onset (57 ± 12.5 years) slowly progressive ataxia with an episodic component in 51% of patients. Balance and gait impairment were almost always present at disease onset. The principal finding on post-mortem examination of 4 brain specimens was loss of Purkinje neurons that was most severe in the vermis most particularly in the anterior vermis. Similar to European populations, a high percent of patients 21/28 (75%) reported a positive treatment response with 4-aminopyridine.
Interpretation
Our study further estimates prevalence and further expands the clinical, imaging and pathological features of SCA27B, while looking at treatment response, disease progression, and survival in patients with this disease. Testing for SCA27B should be considered in all undiagnosed ataxia patients, especially those with episodic onset. ANN NEUROL 2024;96:1092–1103
期刊介绍:
Annals of Neurology publishes original articles with potential for high impact in understanding the pathogenesis, clinical and laboratory features, diagnosis, treatment, outcomes and science underlying diseases of the human nervous system. Articles should ideally be of broad interest to the academic neurological community rather than solely to subspecialists in a particular field. Studies involving experimental model system, including those in cell and organ cultures and animals, of direct translational relevance to the understanding of neurological disease are also encouraged.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
