Chuanhao Dong , Minglin Li , Yanyi Huang , Hai Yang , Bo Wu , Ruoyu Hong
{"title":"旋转电弧等离子体合成石墨烯的分子动力学模拟研究","authors":"Chuanhao Dong , Minglin Li , Yanyi Huang , Hai Yang , Bo Wu , Ruoyu Hong","doi":"10.1016/j.jmgm.2024.108849","DOIUrl":null,"url":null,"abstract":"<div><p>The rotating arc plasma method, based on its unique characteristics, provides a simple, efficient, and catalyst-free approach for graphene material synthesis. This study employs molecular dynamics simulations to theoretically investigate the detailed growth process of graphene at the atomic scale using plasma. During the growth process, different radicals serve as dissociation precursors within the plasma. Simulation results indicate that the growth process of graphene clusters involves three stages: extension of carbon clusters, cyclization of carbon chains, and coalescence of clusters into sheets. Firstly, the precursor concentration affects the size of graphene clusters; increasing the precursor concentration enlarges the cluster size but also increases the likelihood of curling. Secondly, increasing the hydrogen content in the precursor can reduce the growth rate of clusters, decrease dangling bonds at the periphery of clusters, thereby slowing down cluster closure and maintaining a well-defined sheet structure. Lastly, appropriately elevating the simulation temperature can enhance the reaction rate during the simulation process without altering the reaction pathway. These research findings establish the foundation for understanding the growth mechanism of graphene.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108849"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Molecular dynamics simulation study of graphene synthesis by rotating arc plasma\",\"authors\":\"Chuanhao Dong , Minglin Li , Yanyi Huang , Hai Yang , Bo Wu , Ruoyu Hong\",\"doi\":\"10.1016/j.jmgm.2024.108849\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The rotating arc plasma method, based on its unique characteristics, provides a simple, efficient, and catalyst-free approach for graphene material synthesis. This study employs molecular dynamics simulations to theoretically investigate the detailed growth process of graphene at the atomic scale using plasma. During the growth process, different radicals serve as dissociation precursors within the plasma. Simulation results indicate that the growth process of graphene clusters involves three stages: extension of carbon clusters, cyclization of carbon chains, and coalescence of clusters into sheets. Firstly, the precursor concentration affects the size of graphene clusters; increasing the precursor concentration enlarges the cluster size but also increases the likelihood of curling. Secondly, increasing the hydrogen content in the precursor can reduce the growth rate of clusters, decrease dangling bonds at the periphery of clusters, thereby slowing down cluster closure and maintaining a well-defined sheet structure. Lastly, appropriately elevating the simulation temperature can enhance the reaction rate during the simulation process without altering the reaction pathway. These research findings establish the foundation for understanding the growth mechanism of graphene.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"133 \",\"pages\":\"Article 108849\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001499\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001499","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Molecular dynamics simulation study of graphene synthesis by rotating arc plasma
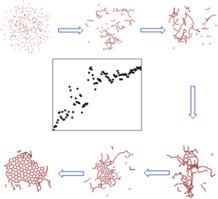
The rotating arc plasma method, based on its unique characteristics, provides a simple, efficient, and catalyst-free approach for graphene material synthesis. This study employs molecular dynamics simulations to theoretically investigate the detailed growth process of graphene at the atomic scale using plasma. During the growth process, different radicals serve as dissociation precursors within the plasma. Simulation results indicate that the growth process of graphene clusters involves three stages: extension of carbon clusters, cyclization of carbon chains, and coalescence of clusters into sheets. Firstly, the precursor concentration affects the size of graphene clusters; increasing the precursor concentration enlarges the cluster size but also increases the likelihood of curling. Secondly, increasing the hydrogen content in the precursor can reduce the growth rate of clusters, decrease dangling bonds at the periphery of clusters, thereby slowing down cluster closure and maintaining a well-defined sheet structure. Lastly, appropriately elevating the simulation temperature can enhance the reaction rate during the simulation process without altering the reaction pathway. These research findings establish the foundation for understanding the growth mechanism of graphene.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
