{"title":"非绝热分子动力学模拟分析中的无监督机器学习","authors":"Yifei Zhu, Jiawei Peng, Chao Xu, Zhenggang Lan","doi":"10.1021/acs.jpclett.4c01751","DOIUrl":null,"url":null,"abstract":"The all-atomic full-dimensional-level simulations of nonadiabatic molecular dynamics (NAMD) in large realistic systems has received high research interest in recent years. However, such NAMD simulations normally generate an enormous amount of time-dependent high-dimensional data, leading to a significant challenge in result analyses. Based on unsupervised machine learning (ML) methods, considerable efforts were devoted to developing novel and easy-to-use analysis tools for the identification of photoinduced reaction channels and the comprehensive understanding of complicated molecular motions in NAMD simulations. Here, we tried to survey recent advances in this field, particularly to focus on how to use unsupervised ML methods to analyze the trajectory-based NAMD simulation results. Our purpose is to offer a comprehensive discussion on several essential components of this analysis protocol, including the selection of ML methods, the construction of molecular descriptors, the establishment of analytical frameworks, their advantages and limitations, and persistent challenges.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"8 1","pages":"9601-9619"},"PeriodicalIF":4.6000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Unsupervised Machine Learning in the Analysis of Nonadiabatic Molecular Dynamics Simulation\",\"authors\":\"Yifei Zhu, Jiawei Peng, Chao Xu, Zhenggang Lan\",\"doi\":\"10.1021/acs.jpclett.4c01751\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The all-atomic full-dimensional-level simulations of nonadiabatic molecular dynamics (NAMD) in large realistic systems has received high research interest in recent years. However, such NAMD simulations normally generate an enormous amount of time-dependent high-dimensional data, leading to a significant challenge in result analyses. Based on unsupervised machine learning (ML) methods, considerable efforts were devoted to developing novel and easy-to-use analysis tools for the identification of photoinduced reaction channels and the comprehensive understanding of complicated molecular motions in NAMD simulations. Here, we tried to survey recent advances in this field, particularly to focus on how to use unsupervised ML methods to analyze the trajectory-based NAMD simulation results. Our purpose is to offer a comprehensive discussion on several essential components of this analysis protocol, including the selection of ML methods, the construction of molecular descriptors, the establishment of analytical frameworks, their advantages and limitations, and persistent challenges.\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"8 1\",\"pages\":\"9601-9619\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-09-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpclett.4c01751\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c01751","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
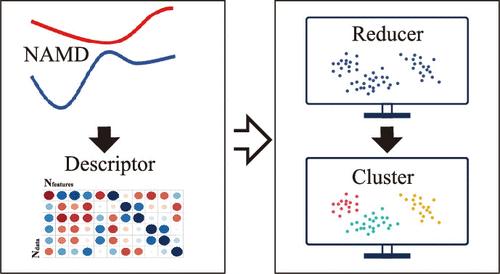
摘要
近年来,大型现实系统中的全原子全维度非绝热分子动力学(NAMD)模拟受到了高度关注。然而,此类 NAMD 模拟通常会产生大量与时间相关的高维数据,给结果分析带来巨大挑战。基于无监督机器学习(ML)方法,人们致力于开发新颖易用的分析工具,用于识别光诱导反应通道和全面理解 NAMD 模拟中复杂的分子运动。在此,我们试图考察这一领域的最新进展,特别是如何使用无监督 ML 方法来分析基于轨迹的 NAMD 模拟结果。我们的目的是全面讨论该分析协议的几个重要组成部分,包括 ML 方法的选择、分子描述符的构建、分析框架的建立、它们的优势和局限性以及持续存在的挑战。
Unsupervised Machine Learning in the Analysis of Nonadiabatic Molecular Dynamics Simulation
The all-atomic full-dimensional-level simulations of nonadiabatic molecular dynamics (NAMD) in large realistic systems has received high research interest in recent years. However, such NAMD simulations normally generate an enormous amount of time-dependent high-dimensional data, leading to a significant challenge in result analyses. Based on unsupervised machine learning (ML) methods, considerable efforts were devoted to developing novel and easy-to-use analysis tools for the identification of photoinduced reaction channels and the comprehensive understanding of complicated molecular motions in NAMD simulations. Here, we tried to survey recent advances in this field, particularly to focus on how to use unsupervised ML methods to analyze the trajectory-based NAMD simulation results. Our purpose is to offer a comprehensive discussion on several essential components of this analysis protocol, including the selection of ML methods, the construction of molecular descriptors, the establishment of analytical frameworks, their advantages and limitations, and persistent challenges.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
