Arseniy A. Otlyotov, Timofey P. Rozov, Andrey D. Moshchenkov, Yury Minenkov
{"title":"16TMCONF543:自动生成的催化相关过渡金属配合物构象能数据集","authors":"Arseniy A. Otlyotov, Timofey P. Rozov, Andrey D. Moshchenkov, Yury Minenkov","doi":"10.1021/acs.organomet.4c00246","DOIUrl":null,"url":null,"abstract":"A database of conformational energies (CEs) of 16 transition metal (TM) complexes relevant to catalysis is automatically created by employing the new conformer generator program Uniconf. The generation procedure starts from an arbitrary high-energy structure and consistently produces conformer(s) similar to or even more energetically favorable than the optimized reference conformer retrieved from the Cambridge Structural Database. The reference CEs obtained with common dispersion-corrected functionals are employed to test the low-cost semiempirical methods. The superiority of the GFN<i>n</i>-xTB schemes over PM6*/7 methods is confirmed by the basic statistical analysis of the CEs. In addition, the influence of the vibrational thermostatistical (Δ<i>G</i><sub>therm</sub>) and continuum solvation (Δ<i>G</i><sub>solv</sub>) corrections on the CEs is examined in the framework of the (modified) scaled rigid-rotor-harmonic oscillator, (m)sRRHO, approximation, and solvation model based on density (SMD). In general, conformational Gibbs free energies exhibit excellent correlation with the respective electronic energies, especially if a more robust msRRHO scheme is employed for the calculation of Δ<i>G</i><sub>therm</sub> and in the case of a nonpolar solvent. The deviations from the perfect correlation occur if reduced CE windows of <5 kcal mol<sup>–1</sup> are considered, implying a greater influence of these effects on the sorting of the low-energy structures.","PeriodicalId":56,"journal":{"name":"Organometallics","volume":"201 1","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"16TMCONF543: An Automatically Generated Data Set of Conformational Energies of Transition Metal Complexes Relevant to Catalysis\",\"authors\":\"Arseniy A. Otlyotov, Timofey P. Rozov, Andrey D. Moshchenkov, Yury Minenkov\",\"doi\":\"10.1021/acs.organomet.4c00246\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"A database of conformational energies (CEs) of 16 transition metal (TM) complexes relevant to catalysis is automatically created by employing the new conformer generator program Uniconf. The generation procedure starts from an arbitrary high-energy structure and consistently produces conformer(s) similar to or even more energetically favorable than the optimized reference conformer retrieved from the Cambridge Structural Database. The reference CEs obtained with common dispersion-corrected functionals are employed to test the low-cost semiempirical methods. The superiority of the GFN<i>n</i>-xTB schemes over PM6*/7 methods is confirmed by the basic statistical analysis of the CEs. In addition, the influence of the vibrational thermostatistical (Δ<i>G</i><sub>therm</sub>) and continuum solvation (Δ<i>G</i><sub>solv</sub>) corrections on the CEs is examined in the framework of the (modified) scaled rigid-rotor-harmonic oscillator, (m)sRRHO, approximation, and solvation model based on density (SMD). In general, conformational Gibbs free energies exhibit excellent correlation with the respective electronic energies, especially if a more robust msRRHO scheme is employed for the calculation of Δ<i>G</i><sub>therm</sub> and in the case of a nonpolar solvent. The deviations from the perfect correlation occur if reduced CE windows of <5 kcal mol<sup>–1</sup> are considered, implying a greater influence of these effects on the sorting of the low-energy structures.\",\"PeriodicalId\":56,\"journal\":{\"name\":\"Organometallics\",\"volume\":\"201 1\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Organometallics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.organomet.4c00246\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organometallics","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.organomet.4c00246","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
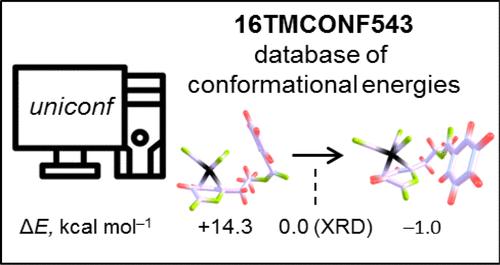
16TMCONF543: An Automatically Generated Data Set of Conformational Energies of Transition Metal Complexes Relevant to Catalysis
A database of conformational energies (CEs) of 16 transition metal (TM) complexes relevant to catalysis is automatically created by employing the new conformer generator program Uniconf. The generation procedure starts from an arbitrary high-energy structure and consistently produces conformer(s) similar to or even more energetically favorable than the optimized reference conformer retrieved from the Cambridge Structural Database. The reference CEs obtained with common dispersion-corrected functionals are employed to test the low-cost semiempirical methods. The superiority of the GFNn-xTB schemes over PM6*/7 methods is confirmed by the basic statistical analysis of the CEs. In addition, the influence of the vibrational thermostatistical (ΔGtherm) and continuum solvation (ΔGsolv) corrections on the CEs is examined in the framework of the (modified) scaled rigid-rotor-harmonic oscillator, (m)sRRHO, approximation, and solvation model based on density (SMD). In general, conformational Gibbs free energies exhibit excellent correlation with the respective electronic energies, especially if a more robust msRRHO scheme is employed for the calculation of ΔGtherm and in the case of a nonpolar solvent. The deviations from the perfect correlation occur if reduced CE windows of <5 kcal mol–1 are considered, implying a greater influence of these effects on the sorting of the low-energy structures.
期刊介绍:
Organometallics is the flagship journal of organometallic chemistry and records progress in one of the most active fields of science, bridging organic and inorganic chemistry. The journal publishes Articles, Communications, Reviews, and Tutorials (instructional overviews) that depict research on the synthesis, structure, bonding, chemical reactivity, and reaction mechanisms for a variety of applications, including catalyst design and catalytic processes; main-group, transition-metal, and lanthanide and actinide metal chemistry; synthetic aspects of polymer science and materials science; and bioorganometallic chemistry.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
