Paolo Surdi, Marina Trivisano, Angela De Dominicis, Mattia Mercier, Ludovica Maria Piscitello, Giusy Carfì Pavia, Costanza Calabrese, Simona Cappelletti, Cinzia Correale, Luigi Mazzone, Federico Vigevano, Nicola Specchio
{"title":"揭示发育性和癫痫性脑病的疾病进展:脑电图和神经心理学的启示","authors":"Paolo Surdi, Marina Trivisano, Angela De Dominicis, Mattia Mercier, Ludovica Maria Piscitello, Giusy Carfì Pavia, Costanza Calabrese, Simona Cappelletti, Cinzia Correale, Luigi Mazzone, Federico Vigevano, Nicola Specchio","doi":"10.1111/epi.18127","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Objective</h3>\n \n <p>Developmental and epileptic encephalopathies (DEEs) are neurological disorders characterized by developmental impairment and epilepsy. Our study aims to assess disease progression by comparing clinical findings, electroencephalography (EEG), and neuropsychological data from seizure onset to the last follow-up evaluation.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>We retrospectively reviewed patients with genetic DEEs who were followed-up at the epilepsy unit of Bambino Gesù Children's Hospital, Rome. We collected information regarding gender, family history, genetic variant, age at onset and at last follow-up, neurological examination, type of seizure, drug resistance, occurrence of status epilepticus, and movement and cognitive and behavioral disorders. We compared EEG background activity, epileptiform abnormalities, and cognitive functions between seizure onset and the last follow-up evaluation using the McNemar-Bowker test (<i>α</i> = 5%).</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>A total of 160 patients (94 female) were included. Genetic analysis revealed a spectrum of pathogenic variants, with <i>SCN1A</i> being the most prevalent (25%). The median age at seizure onset and at the last follow-up was 0.37 (interquartile range [IQR]: 0.09–0.75) and 8.54 years (IQR: 4.32–14.55), respectively. We documented a statistically significant difference in EEG background activity (<i>p</i> = .017) and cognitive impairment (<i>p</i> = .01) from seizure onset to the last follow-up evaluation. No significant differences were detected for epileptiform abnormalities (<i>p</i> = .2). In addition, high prevalence rates were observed for drug resistance (81.9%), movement disorders (60.6%), behavioral and autism spectrum disorders (45%), neurological deficits (31.3%), and occurrence of status epilepticus (23.1%).</p>\n </section>\n \n <section>\n \n <h3> Significance</h3>\n \n <p>Our study provides evidence that a clinical progression may appear in genetic DEEs, manifesting as development or worsening of cognitive impairment and disruption of EEG background activity. These results highlight the challenging clinical course and the importance of early intervention and personalized care in the management of patients with DEEs.</p>\n </section>\n </div>","PeriodicalId":11768,"journal":{"name":"Epilepsia","volume":"65 11","pages":"3279-3292"},"PeriodicalIF":6.6000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/epi.18127","citationCount":"0","resultStr":"{\"title\":\"Unveiling the disease progression in developmental and epileptic encephalopathies: Insights from EEG and neuropsychology\",\"authors\":\"Paolo Surdi, Marina Trivisano, Angela De Dominicis, Mattia Mercier, Ludovica Maria Piscitello, Giusy Carfì Pavia, Costanza Calabrese, Simona Cappelletti, Cinzia Correale, Luigi Mazzone, Federico Vigevano, Nicola Specchio\",\"doi\":\"10.1111/epi.18127\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>Developmental and epileptic encephalopathies (DEEs) are neurological disorders characterized by developmental impairment and epilepsy. Our study aims to assess disease progression by comparing clinical findings, electroencephalography (EEG), and neuropsychological data from seizure onset to the last follow-up evaluation.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>We retrospectively reviewed patients with genetic DEEs who were followed-up at the epilepsy unit of Bambino Gesù Children's Hospital, Rome. We collected information regarding gender, family history, genetic variant, age at onset and at last follow-up, neurological examination, type of seizure, drug resistance, occurrence of status epilepticus, and movement and cognitive and behavioral disorders. We compared EEG background activity, epileptiform abnormalities, and cognitive functions between seizure onset and the last follow-up evaluation using the McNemar-Bowker test (<i>α</i> = 5%).</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>A total of 160 patients (94 female) were included. Genetic analysis revealed a spectrum of pathogenic variants, with <i>SCN1A</i> being the most prevalent (25%). The median age at seizure onset and at the last follow-up was 0.37 (interquartile range [IQR]: 0.09–0.75) and 8.54 years (IQR: 4.32–14.55), respectively. We documented a statistically significant difference in EEG background activity (<i>p</i> = .017) and cognitive impairment (<i>p</i> = .01) from seizure onset to the last follow-up evaluation. No significant differences were detected for epileptiform abnormalities (<i>p</i> = .2). In addition, high prevalence rates were observed for drug resistance (81.9%), movement disorders (60.6%), behavioral and autism spectrum disorders (45%), neurological deficits (31.3%), and occurrence of status epilepticus (23.1%).</p>\\n </section>\\n \\n <section>\\n \\n <h3> Significance</h3>\\n \\n <p>Our study provides evidence that a clinical progression may appear in genetic DEEs, manifesting as development or worsening of cognitive impairment and disruption of EEG background activity. These results highlight the challenging clinical course and the importance of early intervention and personalized care in the management of patients with DEEs.</p>\\n </section>\\n </div>\",\"PeriodicalId\":11768,\"journal\":{\"name\":\"Epilepsia\",\"volume\":\"65 11\",\"pages\":\"3279-3292\"},\"PeriodicalIF\":6.6000,\"publicationDate\":\"2024-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/epi.18127\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Epilepsia\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/epi.18127\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epilepsia","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/epi.18127","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Unveiling the disease progression in developmental and epileptic encephalopathies: Insights from EEG and neuropsychology
Objective
Developmental and epileptic encephalopathies (DEEs) are neurological disorders characterized by developmental impairment and epilepsy. Our study aims to assess disease progression by comparing clinical findings, electroencephalography (EEG), and neuropsychological data from seizure onset to the last follow-up evaluation.
Methods
We retrospectively reviewed patients with genetic DEEs who were followed-up at the epilepsy unit of Bambino Gesù Children's Hospital, Rome. We collected information regarding gender, family history, genetic variant, age at onset and at last follow-up, neurological examination, type of seizure, drug resistance, occurrence of status epilepticus, and movement and cognitive and behavioral disorders. We compared EEG background activity, epileptiform abnormalities, and cognitive functions between seizure onset and the last follow-up evaluation using the McNemar-Bowker test (α = 5%).
Results
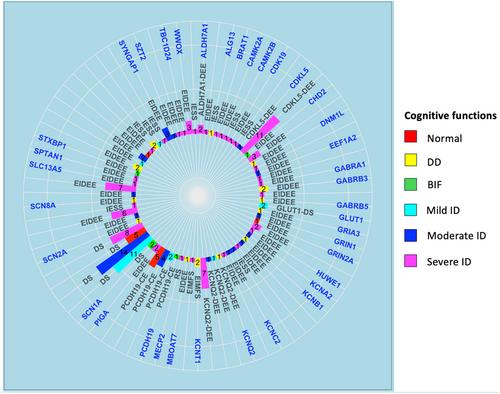
A total of 160 patients (94 female) were included. Genetic analysis revealed a spectrum of pathogenic variants, with SCN1A being the most prevalent (25%). The median age at seizure onset and at the last follow-up was 0.37 (interquartile range [IQR]: 0.09–0.75) and 8.54 years (IQR: 4.32–14.55), respectively. We documented a statistically significant difference in EEG background activity (p = .017) and cognitive impairment (p = .01) from seizure onset to the last follow-up evaluation. No significant differences were detected for epileptiform abnormalities (p = .2). In addition, high prevalence rates were observed for drug resistance (81.9%), movement disorders (60.6%), behavioral and autism spectrum disorders (45%), neurological deficits (31.3%), and occurrence of status epilepticus (23.1%).
Significance
Our study provides evidence that a clinical progression may appear in genetic DEEs, manifesting as development or worsening of cognitive impairment and disruption of EEG background activity. These results highlight the challenging clinical course and the importance of early intervention and personalized care in the management of patients with DEEs.
期刊介绍:
Epilepsia is the leading, authoritative source for innovative clinical and basic science research for all aspects of epilepsy and seizures. In addition, Epilepsia publishes critical reviews, opinion pieces, and guidelines that foster understanding and aim to improve the diagnosis and treatment of people with seizures and epilepsy.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
