{"title":"利用主动深度学习穿越化学空间,实现低数据药物发现。","authors":"Derek van Tilborg, Francesca Grisoni","doi":"10.1038/s43588-024-00697-2","DOIUrl":null,"url":null,"abstract":"Deep learning is accelerating drug discovery. However, current approaches are often affected by limitations in the available data, in terms of either size or molecular diversity. Active deep learning has high potential for low-data drug discovery, as it allows iterative model improvement during the screening process. However, there are several ‘known unknowns’ that limit the wider adoption of active deep learning in drug discovery: (1) what the best computational strategies are for chemical space exploration, (2) how active learning holds up to traditional, non-iterative, approaches and (3) how it should be used in the low-data scenarios typical of drug discovery. To provide answers, this study simulates a low-data drug discovery scenario, and systematically analyzes six active learning strategies combined with two deep learning architectures, on three large-scale molecular libraries. We identify the most important determinants of success in low-data regimes and show that active learning can achieve up to a sixfold improvement in hit discovery when compared with traditional screening methods. Active deep learning is a promising approach to learn from low-data scenarios in drug discovery. This study illuminates key success factors of active learning and shows that it can boost hit discovery by up to sixfold over traditional methods.","PeriodicalId":74246,"journal":{"name":"Nature computational science","volume":"4 10","pages":"786-796"},"PeriodicalIF":20.3000,"publicationDate":"2024-09-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Traversing chemical space with active deep learning for low-data drug discovery\",\"authors\":\"Derek van Tilborg, Francesca Grisoni\",\"doi\":\"10.1038/s43588-024-00697-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Deep learning is accelerating drug discovery. However, current approaches are often affected by limitations in the available data, in terms of either size or molecular diversity. Active deep learning has high potential for low-data drug discovery, as it allows iterative model improvement during the screening process. However, there are several ‘known unknowns’ that limit the wider adoption of active deep learning in drug discovery: (1) what the best computational strategies are for chemical space exploration, (2) how active learning holds up to traditional, non-iterative, approaches and (3) how it should be used in the low-data scenarios typical of drug discovery. To provide answers, this study simulates a low-data drug discovery scenario, and systematically analyzes six active learning strategies combined with two deep learning architectures, on three large-scale molecular libraries. We identify the most important determinants of success in low-data regimes and show that active learning can achieve up to a sixfold improvement in hit discovery when compared with traditional screening methods. Active deep learning is a promising approach to learn from low-data scenarios in drug discovery. This study illuminates key success factors of active learning and shows that it can boost hit discovery by up to sixfold over traditional methods.\",\"PeriodicalId\":74246,\"journal\":{\"name\":\"Nature computational science\",\"volume\":\"4 10\",\"pages\":\"786-796\"},\"PeriodicalIF\":20.3000,\"publicationDate\":\"2024-09-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature computational science\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s43588-024-00697-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature computational science","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43588-024-00697-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
Traversing chemical space with active deep learning for low-data drug discovery
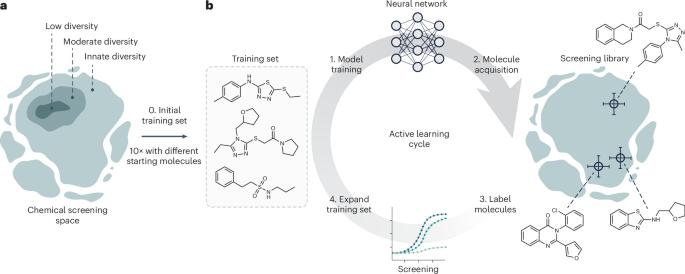
Deep learning is accelerating drug discovery. However, current approaches are often affected by limitations in the available data, in terms of either size or molecular diversity. Active deep learning has high potential for low-data drug discovery, as it allows iterative model improvement during the screening process. However, there are several ‘known unknowns’ that limit the wider adoption of active deep learning in drug discovery: (1) what the best computational strategies are for chemical space exploration, (2) how active learning holds up to traditional, non-iterative, approaches and (3) how it should be used in the low-data scenarios typical of drug discovery. To provide answers, this study simulates a low-data drug discovery scenario, and systematically analyzes six active learning strategies combined with two deep learning architectures, on three large-scale molecular libraries. We identify the most important determinants of success in low-data regimes and show that active learning can achieve up to a sixfold improvement in hit discovery when compared with traditional screening methods. Active deep learning is a promising approach to learn from low-data scenarios in drug discovery. This study illuminates key success factors of active learning and shows that it can boost hit discovery by up to sixfold over traditional methods.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
