{"title":"6-hydroxymethyl-7,8-dihydroptein pyrophosphokinase 的双底物抑制剂:细胞渗透性。","authors":"Genbin Shi , Gary X. Shaw , Xinhua Ji","doi":"10.1016/j.bmcl.2024.129977","DOIUrl":null,"url":null,"abstract":"<div><div>6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) is a key enzyme in the folate biosynthesis pathway. It catalyzes the pyrophosphoryl transfer from ATP to 6-hydroxymethyl-7,8-dihydropterin (HP). HPPK is essential for microorganisms but is absent in mammals. Yet, it is not the target of any existing antibiotics. Hence, this enzyme is an attractive target for developing novel antimicrobial agents. A wealth of structural and mechanistic information has provided solid basis for structure-based design of HPPK inhibitors. Our bisubstrate inhibitors were initially created by linking 6-hydroxymethylpterin to adenosine through 2, 3, or 4 phosphate groups (HP<sub>n</sub>A, n = 2, 3, or 4), among which HP<sub>4</sub>A exhibited the highest binding affinity (K<sub>d</sub> = 0.47 ± 0.04 μM). Further development was carried out based on high-resolution structures of HPPK in complex with HP<sub>4</sub>A. Replacing the phosphate bridge with a piperidine linked thioether eliminated multiple negative charges of the bridge. Substituting the pterin moiety with 7,7-dimethyl-7,8-dihydropterin improved the binding affinity. Arming the piperidine ring with a carboxyl group and oxidizing the thioether further enhanced the potency, resulting in a druglike inhibitor of HPPK (K<sub>d</sub> = 0.047 ± 0.007 μM). None of these inhibitors, however, exhibits bacterial cell permeability. It is most likely due to the lack of active folate transporters in bacteria. Replacing the pterin moiety with a 7-deazagaunine moiety, we have obtained a novel bisubstrate inhibitor (HP-101) showing observable cell permeability toward a Gram-positive bacterium. Here, we report the in vitro activity of HP-101 and its structure in complex with HPPK, providing a framework for structure-based further development.</div></div>","PeriodicalId":256,"journal":{"name":"Bioorganic & Medicinal Chemistry Letters","volume":"113 ","pages":"Article 129977"},"PeriodicalIF":2.2000,"publicationDate":"2024-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Bisubstrate inhibitors of 6-hydroxymethyl-7,8-dihydroptein pyrophosphokinase: Toward cell permeability\",\"authors\":\"Genbin Shi , Gary X. Shaw , Xinhua Ji\",\"doi\":\"10.1016/j.bmcl.2024.129977\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) is a key enzyme in the folate biosynthesis pathway. It catalyzes the pyrophosphoryl transfer from ATP to 6-hydroxymethyl-7,8-dihydropterin (HP). HPPK is essential for microorganisms but is absent in mammals. Yet, it is not the target of any existing antibiotics. Hence, this enzyme is an attractive target for developing novel antimicrobial agents. A wealth of structural and mechanistic information has provided solid basis for structure-based design of HPPK inhibitors. Our bisubstrate inhibitors were initially created by linking 6-hydroxymethylpterin to adenosine through 2, 3, or 4 phosphate groups (HP<sub>n</sub>A, n = 2, 3, or 4), among which HP<sub>4</sub>A exhibited the highest binding affinity (K<sub>d</sub> = 0.47 ± 0.04 μM). Further development was carried out based on high-resolution structures of HPPK in complex with HP<sub>4</sub>A. Replacing the phosphate bridge with a piperidine linked thioether eliminated multiple negative charges of the bridge. Substituting the pterin moiety with 7,7-dimethyl-7,8-dihydropterin improved the binding affinity. Arming the piperidine ring with a carboxyl group and oxidizing the thioether further enhanced the potency, resulting in a druglike inhibitor of HPPK (K<sub>d</sub> = 0.047 ± 0.007 μM). None of these inhibitors, however, exhibits bacterial cell permeability. It is most likely due to the lack of active folate transporters in bacteria. Replacing the pterin moiety with a 7-deazagaunine moiety, we have obtained a novel bisubstrate inhibitor (HP-101) showing observable cell permeability toward a Gram-positive bacterium. Here, we report the in vitro activity of HP-101 and its structure in complex with HPPK, providing a framework for structure-based further development.</div></div>\",\"PeriodicalId\":256,\"journal\":{\"name\":\"Bioorganic & Medicinal Chemistry Letters\",\"volume\":\"113 \",\"pages\":\"Article 129977\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-11-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioorganic & Medicinal Chemistry Letters\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0960894X24003792\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic & Medicinal Chemistry Letters","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0960894X24003792","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/25 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Bisubstrate inhibitors of 6-hydroxymethyl-7,8-dihydroptein pyrophosphokinase: Toward cell permeability
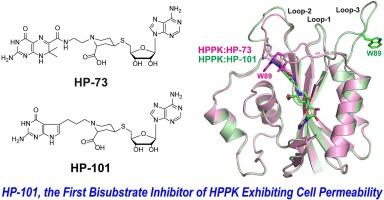
6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) is a key enzyme in the folate biosynthesis pathway. It catalyzes the pyrophosphoryl transfer from ATP to 6-hydroxymethyl-7,8-dihydropterin (HP). HPPK is essential for microorganisms but is absent in mammals. Yet, it is not the target of any existing antibiotics. Hence, this enzyme is an attractive target for developing novel antimicrobial agents. A wealth of structural and mechanistic information has provided solid basis for structure-based design of HPPK inhibitors. Our bisubstrate inhibitors were initially created by linking 6-hydroxymethylpterin to adenosine through 2, 3, or 4 phosphate groups (HPnA, n = 2, 3, or 4), among which HP4A exhibited the highest binding affinity (Kd = 0.47 ± 0.04 μM). Further development was carried out based on high-resolution structures of HPPK in complex with HP4A. Replacing the phosphate bridge with a piperidine linked thioether eliminated multiple negative charges of the bridge. Substituting the pterin moiety with 7,7-dimethyl-7,8-dihydropterin improved the binding affinity. Arming the piperidine ring with a carboxyl group and oxidizing the thioether further enhanced the potency, resulting in a druglike inhibitor of HPPK (Kd = 0.047 ± 0.007 μM). None of these inhibitors, however, exhibits bacterial cell permeability. It is most likely due to the lack of active folate transporters in bacteria. Replacing the pterin moiety with a 7-deazagaunine moiety, we have obtained a novel bisubstrate inhibitor (HP-101) showing observable cell permeability toward a Gram-positive bacterium. Here, we report the in vitro activity of HP-101 and its structure in complex with HPPK, providing a framework for structure-based further development.
期刊介绍:
Bioorganic & Medicinal Chemistry Letters presents preliminary experimental or theoretical research results of outstanding significance and timeliness on all aspects of science at the interface of chemistry and biology and on major advances in drug design and development. The journal publishes articles in the form of communications reporting experimental or theoretical results of special interest, and strives to provide maximum dissemination to a large, international audience.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
