Apurba Nandi, Priyanka Pandey, Paul L Houston, Chen Qu, Qi Yu, Riccardo Conte, Alexandre Tkatchenko, Joel M Bowman
{"title":"通过 Δ 机器学习将基于 DFT 的势能和力场提升到 CCSD(T) 水平的乙醇图解。","authors":"Apurba Nandi, Priyanka Pandey, Paul L Houston, Chen Qu, Qi Yu, Riccardo Conte, Alexandre Tkatchenko, Joel M Bowman","doi":"10.1021/acs.jctc.4c00977","DOIUrl":null,"url":null,"abstract":"<p><p>Progress in machine learning has facilitated the development of potentials that offer both the accuracy of first-principles techniques and vast increases in the speed of evaluation. Recently, Δ-machine learning has been used to elevate the quality of a potential energy surface (PES) based on low-level, e.g., density functional theory (DFT) energies and gradients to close to the gold-standard coupled cluster level of accuracy. We have demonstrated the success of this approach for molecules, ranging in size from H<sub>3</sub>O<sup>+</sup> to 15-atom acetyl-acetone and tropolone. These were all done using the B3LYP functional. Here, we investigate the generality of this approach for the PBE, M06, M06-2X, and PBE0 + MBD functionals, using ethanol as the example molecule. Linear regression with permutationally invariant polynomials is used to fit both low-level and correction PESs. These PESs are employed for standard RMSE analysis for training and test data sets, and then general fidelity tests such as energetics of stationary points, normal-mode frequencies, and torsional potentials are examined. We achieve similar improvements in all cases. Interestingly, we obtained significant improvement over DFT gradients where coupled cluster gradients were not used to correct the low-level PES. Finally, we present some results for correcting a recent molecular mechanics force field for ethanol and comment on the possible generality of this approach.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Δ-Machine Learning to Elevate DFT-Based Potentials and a Force Field to the CCSD(<i>T</i>) Level Illustrated for Ethanol.\",\"authors\":\"Apurba Nandi, Priyanka Pandey, Paul L Houston, Chen Qu, Qi Yu, Riccardo Conte, Alexandre Tkatchenko, Joel M Bowman\",\"doi\":\"10.1021/acs.jctc.4c00977\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Progress in machine learning has facilitated the development of potentials that offer both the accuracy of first-principles techniques and vast increases in the speed of evaluation. Recently, Δ-machine learning has been used to elevate the quality of a potential energy surface (PES) based on low-level, e.g., density functional theory (DFT) energies and gradients to close to the gold-standard coupled cluster level of accuracy. We have demonstrated the success of this approach for molecules, ranging in size from H<sub>3</sub>O<sup>+</sup> to 15-atom acetyl-acetone and tropolone. These were all done using the B3LYP functional. Here, we investigate the generality of this approach for the PBE, M06, M06-2X, and PBE0 + MBD functionals, using ethanol as the example molecule. Linear regression with permutationally invariant polynomials is used to fit both low-level and correction PESs. These PESs are employed for standard RMSE analysis for training and test data sets, and then general fidelity tests such as energetics of stationary points, normal-mode frequencies, and torsional potentials are examined. We achieve similar improvements in all cases. Interestingly, we obtained significant improvement over DFT gradients where coupled cluster gradients were not used to correct the low-level PES. Finally, we present some results for correcting a recent molecular mechanics force field for ethanol and comment on the possible generality of this approach.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-10-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00977\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00977","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
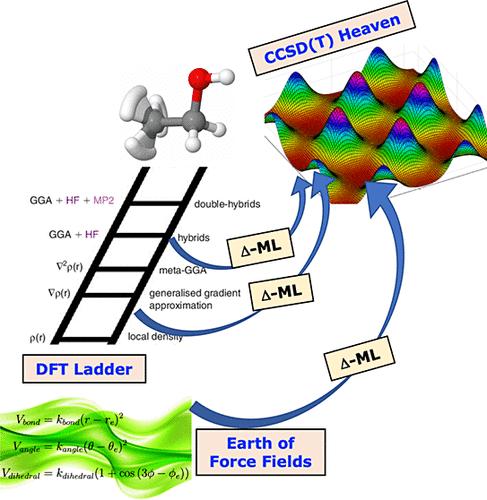
Δ-Machine Learning to Elevate DFT-Based Potentials and a Force Field to the CCSD(T) Level Illustrated for Ethanol.
Progress in machine learning has facilitated the development of potentials that offer both the accuracy of first-principles techniques and vast increases in the speed of evaluation. Recently, Δ-machine learning has been used to elevate the quality of a potential energy surface (PES) based on low-level, e.g., density functional theory (DFT) energies and gradients to close to the gold-standard coupled cluster level of accuracy. We have demonstrated the success of this approach for molecules, ranging in size from H3O+ to 15-atom acetyl-acetone and tropolone. These were all done using the B3LYP functional. Here, we investigate the generality of this approach for the PBE, M06, M06-2X, and PBE0 + MBD functionals, using ethanol as the example molecule. Linear regression with permutationally invariant polynomials is used to fit both low-level and correction PESs. These PESs are employed for standard RMSE analysis for training and test data sets, and then general fidelity tests such as energetics of stationary points, normal-mode frequencies, and torsional potentials are examined. We achieve similar improvements in all cases. Interestingly, we obtained significant improvement over DFT gradients where coupled cluster gradients were not used to correct the low-level PES. Finally, we present some results for correcting a recent molecular mechanics force field for ethanol and comment on the possible generality of this approach.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
