Muhammad Azam, Yibo Chen, Micheal Olaolu Arowolo, Haowang Liu, Mihail Popescu, Dong Xu
{"title":"对挖掘基因关系和路径知识的大型语言模型进行综合评估。","authors":"Muhammad Azam, Yibo Chen, Micheal Olaolu Arowolo, Haowang Liu, Mihail Popescu, Dong Xu","doi":"10.1002/qub2.57","DOIUrl":null,"url":null,"abstract":"<p><p>Understanding complex biological pathways, including gene-gene interactions and gene regulatory networks, is critical for exploring disease mechanisms and drug development. Manual literature curation of biological pathways cannot keep up with the exponential growth of new discoveries in the literature. Large-scale language models (LLMs) trained on extensive text corpora contain rich biological information, and they can be mined as a biological knowledge graph. This study assesses 21 LLMs, including both application programming interface (API)-based models and open-source models in their capacities of retrieving biological knowledge. The evaluation focuses on predicting gene regulatory relations (activation, inhibition, and phosphorylation) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway components. Results indicated a significant disparity in model performance. API-based models GPT-4 and Claude-Pro showed superior performance, with an F1 score of 0.4448 and 0.4386 for the gene regulatory relation prediction, and a Jaccard similarity index of 0.2778 and 0.2657 for the KEGG pathway prediction, respectively. Open-source models lagged behind their API-based counterparts, whereas Falcon-180b and llama2-7b had the highest F1 scores of 0.2787 and 0.1923 in gene regulatory relations, respectively. The KEGG pathway recognition had a Jaccard similarity index of 0.2237 for Falcon-180b and 0.2207 for llama2-7b. Our study suggests that LLMs are informative in gene network analysis and pathway mapping, but their effectiveness varies, necessitating careful model selection. This work also provides a case study and insight into using LLMs das knowledge graphs. Our code is publicly available at the website of GitHub (Muh-aza).</p>","PeriodicalId":45660,"journal":{"name":"Quantitative Biology","volume":"12 4","pages":"360-374"},"PeriodicalIF":1.4000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11446478/pdf/","citationCount":"0","resultStr":"{\"title\":\"A comprehensive evaluation of large language models in mining gene relations and pathway knowledge.\",\"authors\":\"Muhammad Azam, Yibo Chen, Micheal Olaolu Arowolo, Haowang Liu, Mihail Popescu, Dong Xu\",\"doi\":\"10.1002/qub2.57\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Understanding complex biological pathways, including gene-gene interactions and gene regulatory networks, is critical for exploring disease mechanisms and drug development. Manual literature curation of biological pathways cannot keep up with the exponential growth of new discoveries in the literature. Large-scale language models (LLMs) trained on extensive text corpora contain rich biological information, and they can be mined as a biological knowledge graph. This study assesses 21 LLMs, including both application programming interface (API)-based models and open-source models in their capacities of retrieving biological knowledge. The evaluation focuses on predicting gene regulatory relations (activation, inhibition, and phosphorylation) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway components. Results indicated a significant disparity in model performance. API-based models GPT-4 and Claude-Pro showed superior performance, with an F1 score of 0.4448 and 0.4386 for the gene regulatory relation prediction, and a Jaccard similarity index of 0.2778 and 0.2657 for the KEGG pathway prediction, respectively. Open-source models lagged behind their API-based counterparts, whereas Falcon-180b and llama2-7b had the highest F1 scores of 0.2787 and 0.1923 in gene regulatory relations, respectively. The KEGG pathway recognition had a Jaccard similarity index of 0.2237 for Falcon-180b and 0.2207 for llama2-7b. Our study suggests that LLMs are informative in gene network analysis and pathway mapping, but their effectiveness varies, necessitating careful model selection. This work also provides a case study and insight into using LLMs das knowledge graphs. Our code is publicly available at the website of GitHub (Muh-aza).</p>\",\"PeriodicalId\":45660,\"journal\":{\"name\":\"Quantitative Biology\",\"volume\":\"12 4\",\"pages\":\"360-374\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11446478/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Quantitative Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1002/qub2.57\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Quantitative Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1002/qub2.57","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/21 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
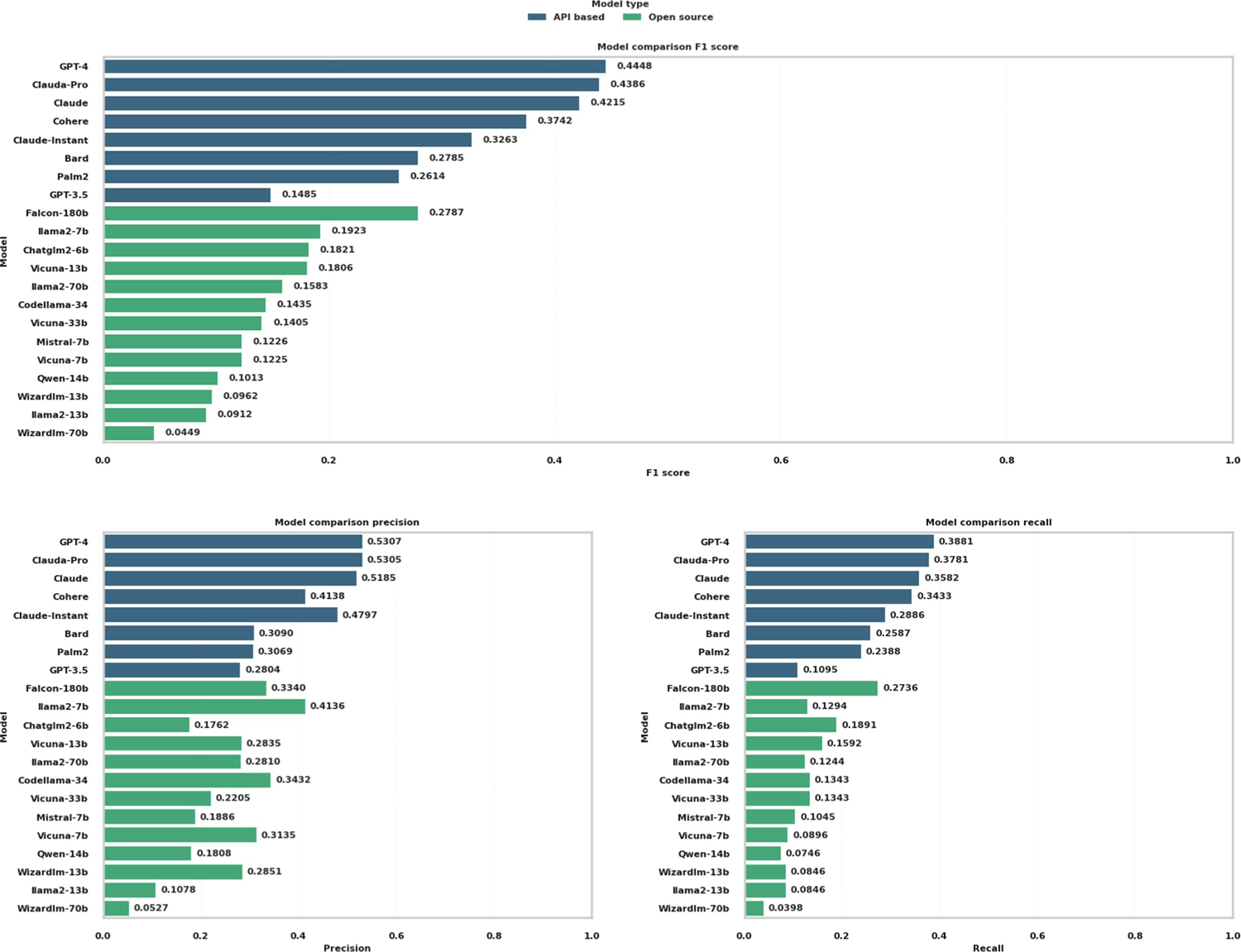
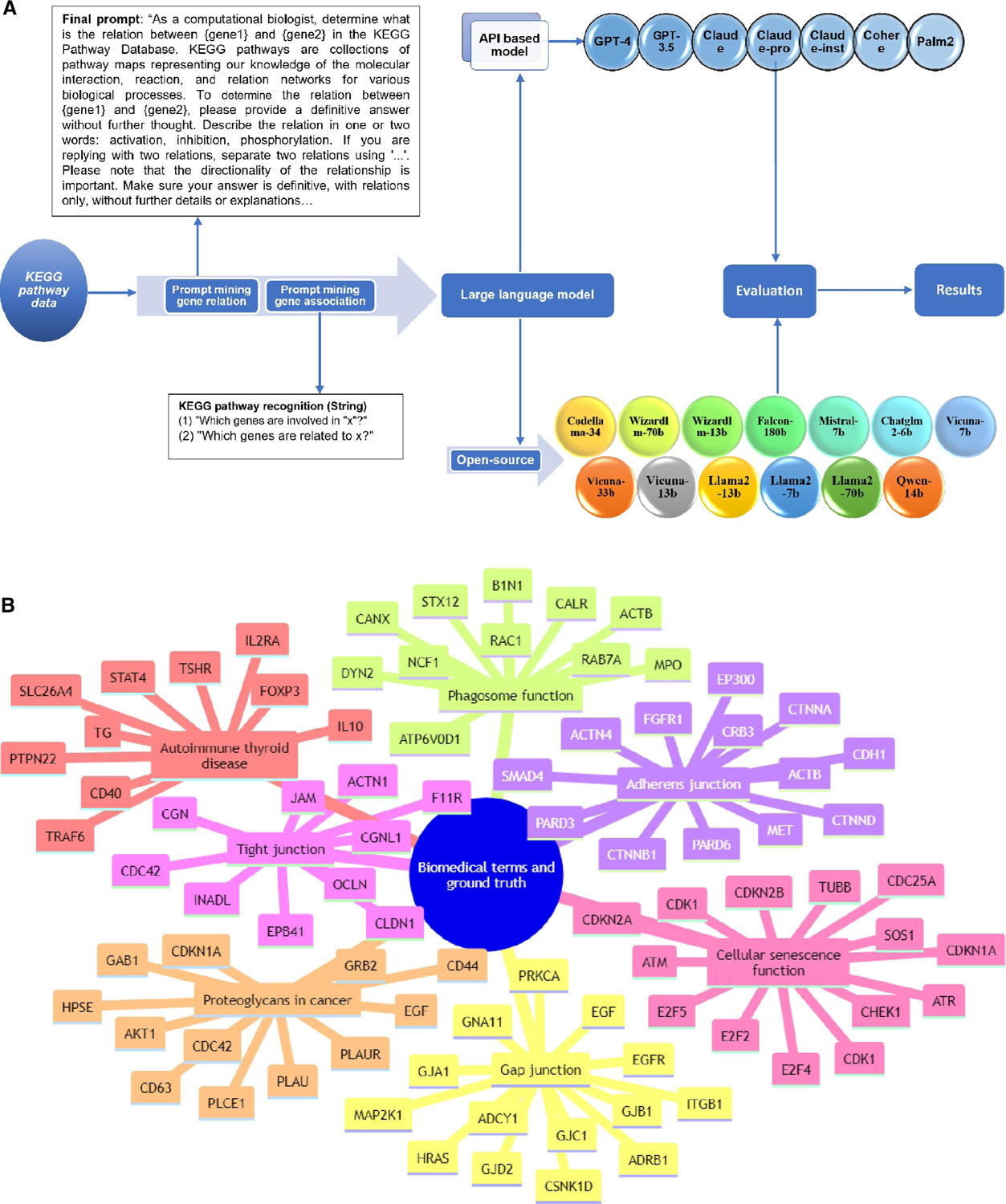
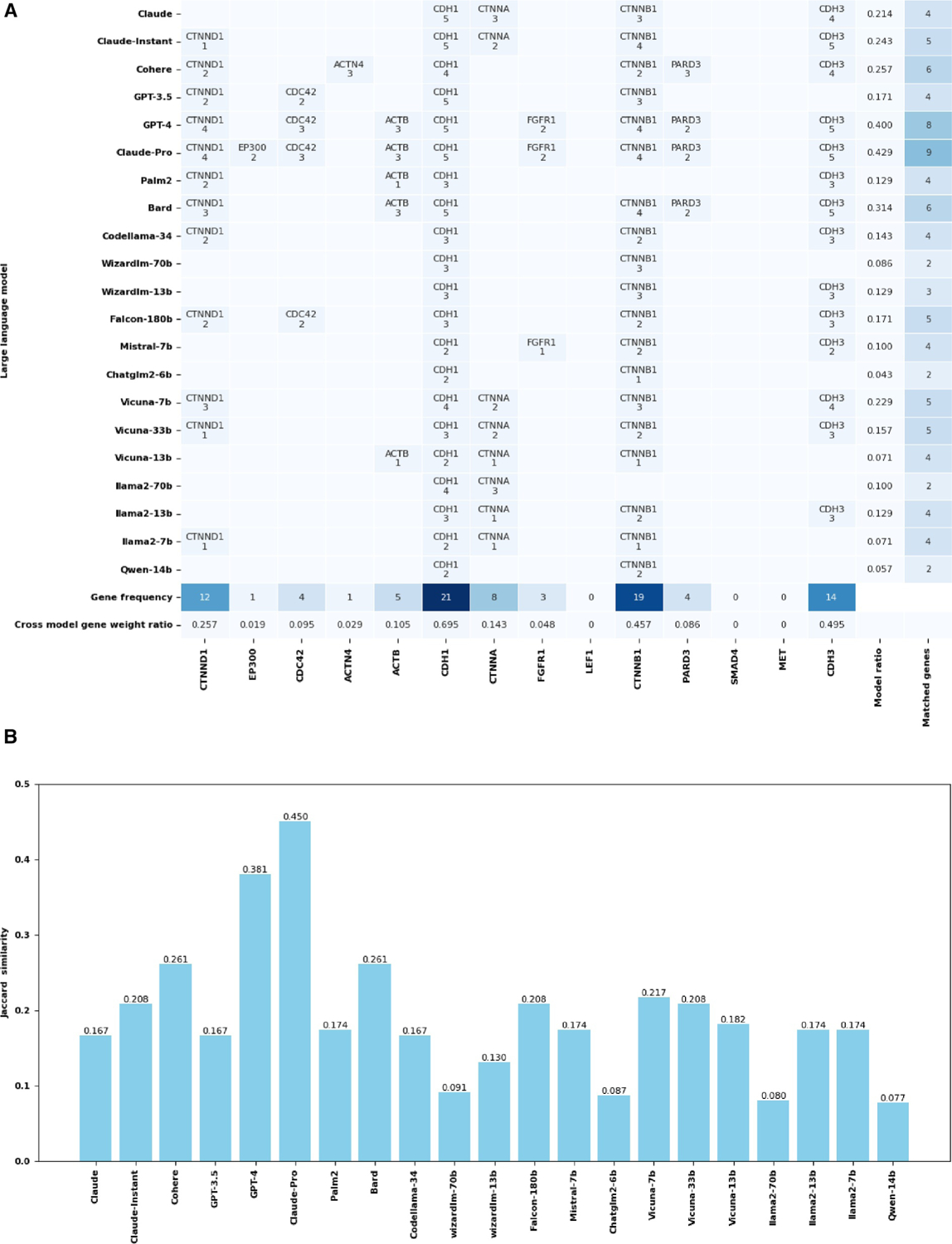
了解复杂的生物通路,包括基因与基因之间的相互作用和基因调控网络,对于探索疾病机理和药物开发至关重要。生物通路的人工文献整理跟不上文献中新发现的指数级增长。在大量文本语料库中训练的大规模语言模型(LLM)包含丰富的生物信息,可以作为生物知识图谱进行挖掘。本研究评估了 21 种 LLM,包括基于应用编程接口(API)的模型和开源模型,以评估它们检索生物知识的能力。评估的重点是预测基因调控关系(激活、抑制和磷酸化)以及《京都基因组百科全书》(KEGG)通路成分。结果表明,模型性能存在明显差异。基于 API 的模型 GPT-4 和 Claude-Pro 表现优异,基因调控关系预测的 F1 分数分别为 0.4448 和 0.4386,KEGG 通路预测的 Jaccard 相似度指数分别为 0.2778 和 0.2657。开源模型落后于基于 API 的模型,而 Falcon-180b 和 llama2-7b 在基因调控关系方面的 F1 分数最高,分别为 0.2787 和 0.1923。在 KEGG 通路识别中,Falcon-180b 和 llama2-7b 的 Jaccard 相似度指数分别为 0.2237 和 0.2207。我们的研究表明,LLMs 在基因网络分析和通路图绘制中具有参考价值,但其有效性各不相同,因此需要谨慎选择模型。这项工作还为使用 LLMs das 知识图谱提供了案例研究和见解。我们的代码可在 GitHub 网站(Muh-aza)上公开获取。
A comprehensive evaluation of large language models in mining gene relations and pathway knowledge.
Understanding complex biological pathways, including gene-gene interactions and gene regulatory networks, is critical for exploring disease mechanisms and drug development. Manual literature curation of biological pathways cannot keep up with the exponential growth of new discoveries in the literature. Large-scale language models (LLMs) trained on extensive text corpora contain rich biological information, and they can be mined as a biological knowledge graph. This study assesses 21 LLMs, including both application programming interface (API)-based models and open-source models in their capacities of retrieving biological knowledge. The evaluation focuses on predicting gene regulatory relations (activation, inhibition, and phosphorylation) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway components. Results indicated a significant disparity in model performance. API-based models GPT-4 and Claude-Pro showed superior performance, with an F1 score of 0.4448 and 0.4386 for the gene regulatory relation prediction, and a Jaccard similarity index of 0.2778 and 0.2657 for the KEGG pathway prediction, respectively. Open-source models lagged behind their API-based counterparts, whereas Falcon-180b and llama2-7b had the highest F1 scores of 0.2787 and 0.1923 in gene regulatory relations, respectively. The KEGG pathway recognition had a Jaccard similarity index of 0.2237 for Falcon-180b and 0.2207 for llama2-7b. Our study suggests that LLMs are informative in gene network analysis and pathway mapping, but their effectiveness varies, necessitating careful model selection. This work also provides a case study and insight into using LLMs das knowledge graphs. Our code is publicly available at the website of GitHub (Muh-aza).
期刊介绍:
Quantitative Biology is an interdisciplinary journal that focuses on original research that uses quantitative approaches and technologies to analyze and integrate biological systems, construct and model engineered life systems, and gain a deeper understanding of the life sciences. It aims to provide a platform for not only the analysis but also the integration and construction of biological systems. It is a quarterly journal seeking to provide an inter- and multi-disciplinary forum for a broad blend of peer-reviewed academic papers in order to promote rapid communication and exchange between scientists in the East and the West. The content of Quantitative Biology will mainly focus on the two broad and related areas: ·bioinformatics and computational biology, which focuses on dealing with information technologies and computational methodologies that can efficiently and accurately manipulate –omics data and transform molecular information into biological knowledge. ·systems and synthetic biology, which focuses on complex interactions in biological systems and the emergent functional properties, and on the design and construction of new biological functions and systems. Its goal is to reflect the significant advances made in quantitatively investigating and modeling both natural and engineered life systems at the molecular and higher levels. The journal particularly encourages original papers that link novel theory with cutting-edge experiments, especially in the newly emerging and multi-disciplinary areas of research. The journal also welcomes high-quality reviews and perspective articles.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
