{"title":"利用 sylph 快速绘制物种级元基因组图谱并估算遏制率","authors":"Jim Shaw, Yun William Yu","doi":"10.1038/s41587-024-02412-y","DOIUrl":null,"url":null,"abstract":"Profiling metagenomes against databases allows for the detection and quantification of microorganisms, even at low abundances where assembly is not possible. We introduce sylph, a species-level metagenome profiler that estimates genome-to-metagenome containment average nucleotide identity (ANI) through zero-inflated Poisson k-mer statistics, enabling ANI-based taxa detection. On the Critical Assessment of Metagenome Interpretation II (CAMI2) Marine dataset, sylph was the most accurate profiling method of seven tested. For multisample profiling, sylph took >10-fold less central processing unit time compared to Kraken2 and used 30-fold less memory. Sylph’s ANI estimates provided an orthogonal signal to abundance, allowing for an ANI-based metagenome-wide association study for Parkinson disease (PD) against 289,232 genomes while confirming known butyrate–PD associations at the strain level. Sylph took <1 min and 16 GB of random-access memory to profile metagenomes against 85,205 prokaryotic and 2,917,516 viral genomes, detecting 30-fold more viral sequences in the human gut compared to RefSeq. Sylph offers precise, efficient profiling with accurate containment ANI estimation even for low-coverage genomes. The microbial composition of metagenomes is identified in seconds by profiling against large databases.","PeriodicalId":19084,"journal":{"name":"Nature biotechnology","volume":"43 8","pages":"1348-1359"},"PeriodicalIF":41.7000,"publicationDate":"2024-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.comhttps://www.nature.com/articles/s41587-024-02412-y.pdf","citationCount":"0","resultStr":"{\"title\":\"Rapid species-level metagenome profiling and containment estimation with sylph\",\"authors\":\"Jim Shaw, Yun William Yu\",\"doi\":\"10.1038/s41587-024-02412-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Profiling metagenomes against databases allows for the detection and quantification of microorganisms, even at low abundances where assembly is not possible. We introduce sylph, a species-level metagenome profiler that estimates genome-to-metagenome containment average nucleotide identity (ANI) through zero-inflated Poisson k-mer statistics, enabling ANI-based taxa detection. On the Critical Assessment of Metagenome Interpretation II (CAMI2) Marine dataset, sylph was the most accurate profiling method of seven tested. For multisample profiling, sylph took >10-fold less central processing unit time compared to Kraken2 and used 30-fold less memory. Sylph’s ANI estimates provided an orthogonal signal to abundance, allowing for an ANI-based metagenome-wide association study for Parkinson disease (PD) against 289,232 genomes while confirming known butyrate–PD associations at the strain level. Sylph took <1 min and 16 GB of random-access memory to profile metagenomes against 85,205 prokaryotic and 2,917,516 viral genomes, detecting 30-fold more viral sequences in the human gut compared to RefSeq. Sylph offers precise, efficient profiling with accurate containment ANI estimation even for low-coverage genomes. The microbial composition of metagenomes is identified in seconds by profiling against large databases.\",\"PeriodicalId\":19084,\"journal\":{\"name\":\"Nature biotechnology\",\"volume\":\"43 8\",\"pages\":\"1348-1359\"},\"PeriodicalIF\":41.7000,\"publicationDate\":\"2024-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.comhttps://www.nature.com/articles/s41587-024-02412-y.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature biotechnology\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://www.nature.com/articles/s41587-024-02412-y\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature biotechnology","FirstCategoryId":"5","ListUrlMain":"https://www.nature.com/articles/s41587-024-02412-y","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Rapid species-level metagenome profiling and containment estimation with sylph
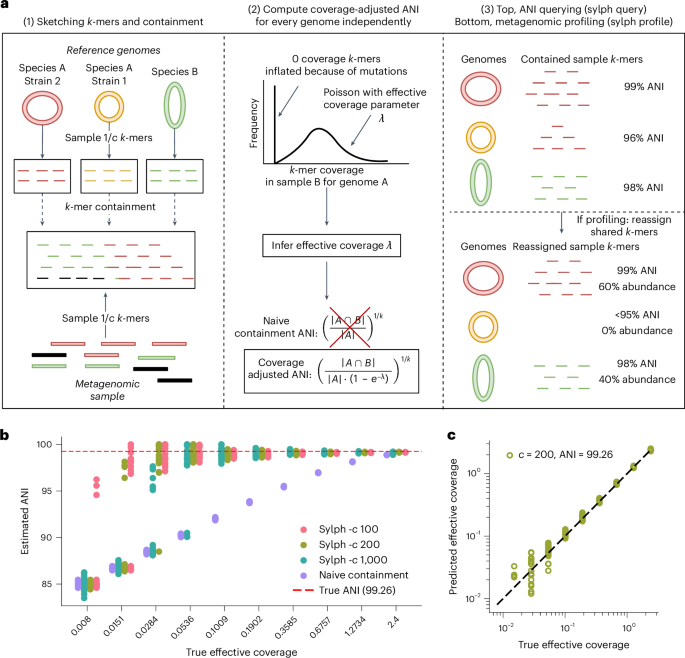
Profiling metagenomes against databases allows for the detection and quantification of microorganisms, even at low abundances where assembly is not possible. We introduce sylph, a species-level metagenome profiler that estimates genome-to-metagenome containment average nucleotide identity (ANI) through zero-inflated Poisson k-mer statistics, enabling ANI-based taxa detection. On the Critical Assessment of Metagenome Interpretation II (CAMI2) Marine dataset, sylph was the most accurate profiling method of seven tested. For multisample profiling, sylph took >10-fold less central processing unit time compared to Kraken2 and used 30-fold less memory. Sylph’s ANI estimates provided an orthogonal signal to abundance, allowing for an ANI-based metagenome-wide association study for Parkinson disease (PD) against 289,232 genomes while confirming known butyrate–PD associations at the strain level. Sylph took <1 min and 16 GB of random-access memory to profile metagenomes against 85,205 prokaryotic and 2,917,516 viral genomes, detecting 30-fold more viral sequences in the human gut compared to RefSeq. Sylph offers precise, efficient profiling with accurate containment ANI estimation even for low-coverage genomes. The microbial composition of metagenomes is identified in seconds by profiling against large databases.
期刊介绍:
Nature Biotechnology is a monthly journal that focuses on the science and business of biotechnology. It covers a wide range of topics including technology/methodology advancements in the biological, biomedical, agricultural, and environmental sciences. The journal also explores the commercial, political, ethical, legal, and societal aspects of this research.
The journal serves researchers by providing peer-reviewed research papers in the field of biotechnology. It also serves the business community by delivering news about research developments. This approach ensures that both the scientific and business communities are well-informed and able to stay up-to-date on the latest advancements and opportunities in the field.
Some key areas of interest in which the journal actively seeks research papers include molecular engineering of nucleic acids and proteins, molecular therapy, large-scale biology, computational biology, regenerative medicine, imaging technology, analytical biotechnology, applied immunology, food and agricultural biotechnology, and environmental biotechnology.
In summary, Nature Biotechnology is a comprehensive journal that covers both the scientific and business aspects of biotechnology. It strives to provide researchers with valuable research papers and news while also delivering important scientific advancements to the business community.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
