Ayush Gupta, Heng Ma, Arvind Ramanathan, Gül H. Zerze
{"title":"探索 RNA 干环相位空间的深度学习驱动采样技术","authors":"Ayush Gupta, Heng Ma, Arvind Ramanathan, Gül H. Zerze","doi":"10.1021/acs.jctc.4c00669","DOIUrl":null,"url":null,"abstract":"The folding and unfolding of RNA stem-loops are critical biological processes; however, their computational studies are often hampered by the ruggedness of their folding landscape, necessitating long simulation times at the atomistic scale. Here, we adapted DeepDriveMD (DDMD), an advanced deep learning-driven sampling technique originally developed for protein folding, to address the challenges of RNA stem-loop folding. Although tempering- and order parameter-based techniques are commonly used for similar rare-event problems, the computational costs or the need for a priori knowledge about the system often present a challenge in their effective use. DDMD overcomes these challenges by adaptively learning from an ensemble of running MD simulations using generic contact maps as the raw input. DeepDriveMD enables on-the-fly learning of a low-dimensional latent representation and guides the simulation toward the undersampled regions while optimizing the resources to explore the relevant parts of the phase space. We showed that DDMD estimates the free energy landscape of the RNA stem-loop reasonably well at room temperature. Our simulation framework runs at a constant temperature without external biasing potential, hence preserving the information on transition rates, with a computational cost much lower than that of the simulations performed with external biasing potentials. We also introduced a reweighting strategy for obtaining unbiased free energy surfaces and presented a qualitative analysis of the latent space. This analysis showed that the latent space captures the relevant slow degrees of freedom for the RNA folding problem of interest. Finally, throughout the manuscript, we outlined how different parameters are selected and optimized to adapt DDMD for this system. We believe this compendium of decision-making processes will help new users adapt this technique for the rare-event sampling problems of their interest.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A Deep Learning-Driven Sampling Technique to Explore the Phase Space of an RNA Stem-Loop\",\"authors\":\"Ayush Gupta, Heng Ma, Arvind Ramanathan, Gül H. Zerze\",\"doi\":\"10.1021/acs.jctc.4c00669\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The folding and unfolding of RNA stem-loops are critical biological processes; however, their computational studies are often hampered by the ruggedness of their folding landscape, necessitating long simulation times at the atomistic scale. Here, we adapted DeepDriveMD (DDMD), an advanced deep learning-driven sampling technique originally developed for protein folding, to address the challenges of RNA stem-loop folding. Although tempering- and order parameter-based techniques are commonly used for similar rare-event problems, the computational costs or the need for a priori knowledge about the system often present a challenge in their effective use. DDMD overcomes these challenges by adaptively learning from an ensemble of running MD simulations using generic contact maps as the raw input. DeepDriveMD enables on-the-fly learning of a low-dimensional latent representation and guides the simulation toward the undersampled regions while optimizing the resources to explore the relevant parts of the phase space. We showed that DDMD estimates the free energy landscape of the RNA stem-loop reasonably well at room temperature. Our simulation framework runs at a constant temperature without external biasing potential, hence preserving the information on transition rates, with a computational cost much lower than that of the simulations performed with external biasing potentials. We also introduced a reweighting strategy for obtaining unbiased free energy surfaces and presented a qualitative analysis of the latent space. This analysis showed that the latent space captures the relevant slow degrees of freedom for the RNA folding problem of interest. Finally, throughout the manuscript, we outlined how different parameters are selected and optimized to adapt DDMD for this system. We believe this compendium of decision-making processes will help new users adapt this technique for the rare-event sampling problems of their interest.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00669\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00669","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A Deep Learning-Driven Sampling Technique to Explore the Phase Space of an RNA Stem-Loop
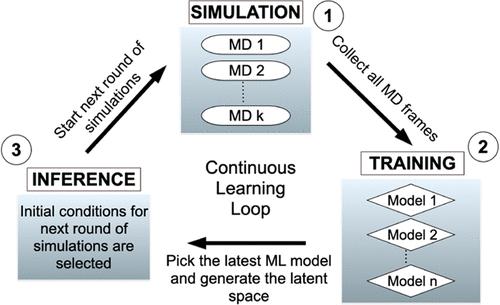
The folding and unfolding of RNA stem-loops are critical biological processes; however, their computational studies are often hampered by the ruggedness of their folding landscape, necessitating long simulation times at the atomistic scale. Here, we adapted DeepDriveMD (DDMD), an advanced deep learning-driven sampling technique originally developed for protein folding, to address the challenges of RNA stem-loop folding. Although tempering- and order parameter-based techniques are commonly used for similar rare-event problems, the computational costs or the need for a priori knowledge about the system often present a challenge in their effective use. DDMD overcomes these challenges by adaptively learning from an ensemble of running MD simulations using generic contact maps as the raw input. DeepDriveMD enables on-the-fly learning of a low-dimensional latent representation and guides the simulation toward the undersampled regions while optimizing the resources to explore the relevant parts of the phase space. We showed that DDMD estimates the free energy landscape of the RNA stem-loop reasonably well at room temperature. Our simulation framework runs at a constant temperature without external biasing potential, hence preserving the information on transition rates, with a computational cost much lower than that of the simulations performed with external biasing potentials. We also introduced a reweighting strategy for obtaining unbiased free energy surfaces and presented a qualitative analysis of the latent space. This analysis showed that the latent space captures the relevant slow degrees of freedom for the RNA folding problem of interest. Finally, throughout the manuscript, we outlined how different parameters are selected and optimized to adapt DDMD for this system. We believe this compendium of decision-making processes will help new users adapt this technique for the rare-event sampling problems of their interest.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
