Matthew R. Hennefarth, Donald G. Truhlar, Laura Gagliardi
{"title":"使用线性化对密度函数理论的半经典非绝热分子动力学","authors":"Matthew R. Hennefarth, Donald G. Truhlar, Laura Gagliardi","doi":"10.1021/acs.jctc.4c01061","DOIUrl":null,"url":null,"abstract":"Nonadiabatic molecular dynamics is an effective method for modeling nonradiative decay in electronically excited molecules. Its accuracy depends strongly on the quality of the potential energy surfaces, and its affordability for long direct-dynamic simulations with adequate ensemble averaging depends strongly on the cost of the required electronic structure calculations. Linearized pair-density functional theory (L-PDFT) is a recently developed post-self-consistent-field multireference method that can model potential energy surfaces with an accuracy similar to expensive multireference perturbation theories but at a computational cost similar to the underlying multiconfiguration self-consistent field method. Here, we integrate the <span>SHARC</span> dynamics and <span>PySCF</span> electronic structure code to utilize L-PDFT for electronically nonadiabatic calculations and use the combined programs to study the photoisomerization reaction of <i>cis</i>-azomethane. We show that L-PDFT is able to successfully simulate the photoisomerization without crashes, and it yields results similar to the more expensive extended multistate complete active space second-order perturbation theory. This shows that L-PDFT can model internal conversion, and it demonstrates its promise for broader photodynamics applications.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"53 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Semiclassical Nonadiabatic Molecular Dynamics Using Linearized Pair-Density Functional Theory\",\"authors\":\"Matthew R. Hennefarth, Donald G. Truhlar, Laura Gagliardi\",\"doi\":\"10.1021/acs.jctc.4c01061\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Nonadiabatic molecular dynamics is an effective method for modeling nonradiative decay in electronically excited molecules. Its accuracy depends strongly on the quality of the potential energy surfaces, and its affordability for long direct-dynamic simulations with adequate ensemble averaging depends strongly on the cost of the required electronic structure calculations. Linearized pair-density functional theory (L-PDFT) is a recently developed post-self-consistent-field multireference method that can model potential energy surfaces with an accuracy similar to expensive multireference perturbation theories but at a computational cost similar to the underlying multiconfiguration self-consistent field method. Here, we integrate the <span>SHARC</span> dynamics and <span>PySCF</span> electronic structure code to utilize L-PDFT for electronically nonadiabatic calculations and use the combined programs to study the photoisomerization reaction of <i>cis</i>-azomethane. We show that L-PDFT is able to successfully simulate the photoisomerization without crashes, and it yields results similar to the more expensive extended multistate complete active space second-order perturbation theory. This shows that L-PDFT can model internal conversion, and it demonstrates its promise for broader photodynamics applications.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"53 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-10-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c01061\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01061","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Semiclassical Nonadiabatic Molecular Dynamics Using Linearized Pair-Density Functional Theory
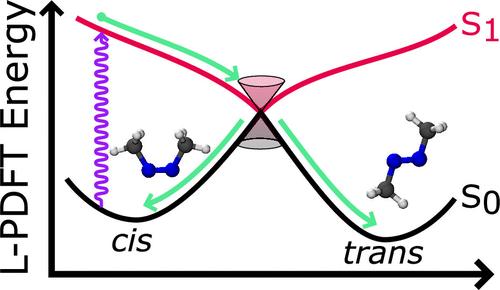
Nonadiabatic molecular dynamics is an effective method for modeling nonradiative decay in electronically excited molecules. Its accuracy depends strongly on the quality of the potential energy surfaces, and its affordability for long direct-dynamic simulations with adequate ensemble averaging depends strongly on the cost of the required electronic structure calculations. Linearized pair-density functional theory (L-PDFT) is a recently developed post-self-consistent-field multireference method that can model potential energy surfaces with an accuracy similar to expensive multireference perturbation theories but at a computational cost similar to the underlying multiconfiguration self-consistent field method. Here, we integrate the SHARC dynamics and PySCF electronic structure code to utilize L-PDFT for electronically nonadiabatic calculations and use the combined programs to study the photoisomerization reaction of cis-azomethane. We show that L-PDFT is able to successfully simulate the photoisomerization without crashes, and it yields results similar to the more expensive extended multistate complete active space second-order perturbation theory. This shows that L-PDFT can model internal conversion, and it demonstrates its promise for broader photodynamics applications.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
