Satadeep Bhattacharjee, Namitha Anna Koshi and Seung-Cheol Lee
{"title":"定制 PBE 交换相关函数:预测各种半导体带隙的综合方法。","authors":"Satadeep Bhattacharjee, Namitha Anna Koshi and Seung-Cheol Lee","doi":"10.1039/D4CP03260H","DOIUrl":null,"url":null,"abstract":"<p >Accurate band gap prediction in semiconductors is crucial for materials science and semiconductor technology advancements. This paper extends the Perdew–Burke–Ernzerhof (PBE) functional for a wide range of semiconductors, tackling the exchange and correlation enhancement factor complexities within density functional theory (DFT). Our customized functionals offer a clearer and more realistic alternative to DFT+<em>U</em> methods, which demand large negative <em>U</em> values for elements like sulfur (S), selenium (Se), and phosphorus (P). Moreover, these functionals are more cost-effective than GW or Heyd–Scuseria–Ernzerhof (HSE) hybrid functional methods, therefore, significantly facilitating the way for unified workflows in analyzing electronic structure, dielectric constants, effective masses, and further transport and elastic properties, allowing for seamless calculations across various properties. We point out that such development could be helpful in the creation of comprehensive databases of band gap and dielectric properties of the materials without expensive calculations. Furthermore, for the semiconductors studied, we show that these customized functionals and the strongly constrained and appropriately normed semilocal density functional (SCAN) perform similarly in terms of the band gap.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 41","pages":" 26443-26452"},"PeriodicalIF":2.9000,"publicationDate":"2024-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Customizing PBE exchange–correlation functionals: a comprehensive approach for band gap prediction in diverse semiconductors†\",\"authors\":\"Satadeep Bhattacharjee, Namitha Anna Koshi and Seung-Cheol Lee\",\"doi\":\"10.1039/D4CP03260H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Accurate band gap prediction in semiconductors is crucial for materials science and semiconductor technology advancements. This paper extends the Perdew–Burke–Ernzerhof (PBE) functional for a wide range of semiconductors, tackling the exchange and correlation enhancement factor complexities within density functional theory (DFT). Our customized functionals offer a clearer and more realistic alternative to DFT+<em>U</em> methods, which demand large negative <em>U</em> values for elements like sulfur (S), selenium (Se), and phosphorus (P). Moreover, these functionals are more cost-effective than GW or Heyd–Scuseria–Ernzerhof (HSE) hybrid functional methods, therefore, significantly facilitating the way for unified workflows in analyzing electronic structure, dielectric constants, effective masses, and further transport and elastic properties, allowing for seamless calculations across various properties. We point out that such development could be helpful in the creation of comprehensive databases of band gap and dielectric properties of the materials without expensive calculations. Furthermore, for the semiconductors studied, we show that these customized functionals and the strongly constrained and appropriately normed semilocal density functional (SCAN) perform similarly in terms of the band gap.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 41\",\"pages\":\" 26443-26452\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp03260h\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp03260h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Customizing PBE exchange–correlation functionals: a comprehensive approach for band gap prediction in diverse semiconductors†
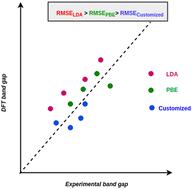
Accurate band gap prediction in semiconductors is crucial for materials science and semiconductor technology advancements. This paper extends the Perdew–Burke–Ernzerhof (PBE) functional for a wide range of semiconductors, tackling the exchange and correlation enhancement factor complexities within density functional theory (DFT). Our customized functionals offer a clearer and more realistic alternative to DFT+U methods, which demand large negative U values for elements like sulfur (S), selenium (Se), and phosphorus (P). Moreover, these functionals are more cost-effective than GW or Heyd–Scuseria–Ernzerhof (HSE) hybrid functional methods, therefore, significantly facilitating the way for unified workflows in analyzing electronic structure, dielectric constants, effective masses, and further transport and elastic properties, allowing for seamless calculations across various properties. We point out that such development could be helpful in the creation of comprehensive databases of band gap and dielectric properties of the materials without expensive calculations. Furthermore, for the semiconductors studied, we show that these customized functionals and the strongly constrained and appropriately normed semilocal density functional (SCAN) perform similarly in terms of the band gap.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
