Huseyin Tunc, Sumeyye Yilmaz, Busra Nur Darendeli Kiraz, Murat Sari, Seyfullah Enes Kotil, Ozge Sensoy, Serdar Durdagi
{"title":"通过迁移学习机制提高新型 HIV-1 蛋白酶抑制剂的抗药性预测功效","authors":"Huseyin Tunc, Sumeyye Yilmaz, Busra Nur Darendeli Kiraz, Murat Sari, Seyfullah Enes Kotil, Ozge Sensoy, Serdar Durdagi","doi":"10.1021/acs.jcim.4c01037","DOIUrl":null,"url":null,"abstract":"<p><p>The human immunodeficiency virus presents a significant global health challenge due to its rapid mutation and the development of resistance mechanisms against antiretroviral drugs. Recent studies demonstrate the impressive performance of machine learning (ML) and deep learning (DL) models in predicting the drug resistance profile of specific FDA-approved inhibitors. However, generalizing ML and DL models to learn not only from isolates but also from inhibitor representations remains challenging for HIV-1 infection. We propose a novel drug-isolate-fold change (DIF) model framework that aims to predict drug resistance score directly from the protein sequence and inhibitor representation. Various ML and DL models, inhibitor representations, and protein representations were analyzed through realistic validation mechanisms. To enhance the molecular learning capacity of DIF models, we employ a transfer learning approach by pretraining a graph neural network (GNN) model for activity prediction on a data set of 4855 HIV-1 protease inhibitors (PIs). By performing various realistic validation strategies on internal and external genotype-phenotype data sets, we statistically show that the learned representations of inhibitors improve the predictive ability of DIF-based ML and DL models. We achieved an accuracy of 0.802, AUROC of 0.874, and <i>r</i> of 0.727 for the unseen external PIs. By comparing the DIF-based models with a null model consisting of isolate-fold change (IF) architecture, it is observed that the DIF models significantly benefit from molecular representations. Combined results from various testing strategies and statistical tests confirm the effectiveness of DIF models in testing novel PIs for drug resistance in the presence of an isolate.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"7844-7863"},"PeriodicalIF":6.4000,"publicationDate":"2024-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Improving Predictive Efficacy for Drug Resistance in Novel HIV-1 Protease Inhibitors through Transfer Learning Mechanisms.\",\"authors\":\"Huseyin Tunc, Sumeyye Yilmaz, Busra Nur Darendeli Kiraz, Murat Sari, Seyfullah Enes Kotil, Ozge Sensoy, Serdar Durdagi\",\"doi\":\"10.1021/acs.jcim.4c01037\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The human immunodeficiency virus presents a significant global health challenge due to its rapid mutation and the development of resistance mechanisms against antiretroviral drugs. Recent studies demonstrate the impressive performance of machine learning (ML) and deep learning (DL) models in predicting the drug resistance profile of specific FDA-approved inhibitors. However, generalizing ML and DL models to learn not only from isolates but also from inhibitor representations remains challenging for HIV-1 infection. We propose a novel drug-isolate-fold change (DIF) model framework that aims to predict drug resistance score directly from the protein sequence and inhibitor representation. Various ML and DL models, inhibitor representations, and protein representations were analyzed through realistic validation mechanisms. To enhance the molecular learning capacity of DIF models, we employ a transfer learning approach by pretraining a graph neural network (GNN) model for activity prediction on a data set of 4855 HIV-1 protease inhibitors (PIs). By performing various realistic validation strategies on internal and external genotype-phenotype data sets, we statistically show that the learned representations of inhibitors improve the predictive ability of DIF-based ML and DL models. We achieved an accuracy of 0.802, AUROC of 0.874, and <i>r</i> of 0.727 for the unseen external PIs. By comparing the DIF-based models with a null model consisting of isolate-fold change (IF) architecture, it is observed that the DIF models significantly benefit from molecular representations. Combined results from various testing strategies and statistical tests confirm the effectiveness of DIF models in testing novel PIs for drug resistance in the presence of an isolate.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"7844-7863\"},\"PeriodicalIF\":6.4000,\"publicationDate\":\"2024-10-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c01037\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01037","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
摘要
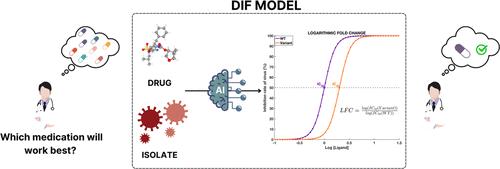
由于人类免疫缺陷病毒的快速变异和抗逆转录病毒药物耐药性机制的发展,人类免疫缺陷病毒给全球健康带来了重大挑战。最近的研究表明,机器学习(ML)和深度学习(DL)模型在预测美国食品及药物管理局(FDA)批准的特定抑制剂的耐药性特征方面表现出色。然而,将 ML 和 DL 模型推广到不仅从分离株而且从抑制剂表征中进行学习,对于 HIV-1 感染来说仍然具有挑战性。我们提出了一种新的药物-分离物-折变(DIF)模型框架,旨在直接从蛋白质序列和抑制剂表征预测耐药性得分。我们通过现实验证机制分析了各种 ML 和 DL 模型、抑制剂表征和蛋白质表征。为了提高 DIF 模型的分子学习能力,我们采用了迁移学习方法,在 4855 种 HIV-1 蛋白酶抑制剂(PIs)数据集上预训练了一个图神经网络(GNN)模型,用于活性预测。通过在内部和外部基因型-表型数据集上执行各种实际验证策略,我们从统计学角度证明,学习到的抑制剂表征提高了基于 DIF 的 ML 和 DL 模型的预测能力。对于未见过的外部 PI,我们的准确率达到了 0.802,AUROC 为 0.874,r 为 0.727。通过将基于 DIF 的模型与由隔离折半变化(IF)结构组成的无效模型进行比较,我们发现 DIF 模型明显受益于分子表征。各种测试策略和统计检验的综合结果证实了 DIF 模型在测试存在分离株的新型 PIs 耐药性方面的有效性。
Improving Predictive Efficacy for Drug Resistance in Novel HIV-1 Protease Inhibitors through Transfer Learning Mechanisms.
The human immunodeficiency virus presents a significant global health challenge due to its rapid mutation and the development of resistance mechanisms against antiretroviral drugs. Recent studies demonstrate the impressive performance of machine learning (ML) and deep learning (DL) models in predicting the drug resistance profile of specific FDA-approved inhibitors. However, generalizing ML and DL models to learn not only from isolates but also from inhibitor representations remains challenging for HIV-1 infection. We propose a novel drug-isolate-fold change (DIF) model framework that aims to predict drug resistance score directly from the protein sequence and inhibitor representation. Various ML and DL models, inhibitor representations, and protein representations were analyzed through realistic validation mechanisms. To enhance the molecular learning capacity of DIF models, we employ a transfer learning approach by pretraining a graph neural network (GNN) model for activity prediction on a data set of 4855 HIV-1 protease inhibitors (PIs). By performing various realistic validation strategies on internal and external genotype-phenotype data sets, we statistically show that the learned representations of inhibitors improve the predictive ability of DIF-based ML and DL models. We achieved an accuracy of 0.802, AUROC of 0.874, and r of 0.727 for the unseen external PIs. By comparing the DIF-based models with a null model consisting of isolate-fold change (IF) architecture, it is observed that the DIF models significantly benefit from molecular representations. Combined results from various testing strategies and statistical tests confirm the effectiveness of DIF models in testing novel PIs for drug resistance in the presence of an isolate.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
