{"title":"对蛋白质二级结构独特特征的统计分析。","authors":"Nitin Kumar Singh , Manish Agarwal , Mithun Radhakrishna","doi":"10.1016/j.compbiolchem.2024.108237","DOIUrl":null,"url":null,"abstract":"<div><div>Protein folding is a complex process influenced by the primary sequence of amino acids. Early studies focused on understanding whether the specificity or the conservation of properties of amino acids was crucial for folding into secondary structures such as <span><math><mi>α</mi></math></span>-helices, <span><math><mi>β</mi></math></span>-sheets, turns, and coils. However, with the advent of artificial intelligence (AI) and machine learning (ML), the emphasis has shifted towards the precise nature and occurrence of specific amino acids. In our study, we analyzed a large set of proteins from diverse organisms to identify unique features of secondary structures, particularly in terms of the distribution of polar, non-polar, and charged amino acid residues. We found that <span><math><mi>α</mi></math></span>-helices tend to have a higher proportion of charged and non-polar groups compared to other secondary structures and that the presence of oppositely charged amino acid residues in helices stabilizes them, facilitating the formation of longer helices. These characteristics are distinct to <span><math><mi>α</mi></math></span>-helices. This study offers valuable insights for researchers in the field of protein design, enabling the de-novo creation of short helical peptides for a range of applications. We have also developed a web server for extensive analysis of proteins from different databases. The web server is housed at <span><span>https://proseqanalyser.iitgn.ac.in/</span><svg><path></path></svg></span></div></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"113 ","pages":"Article 108237"},"PeriodicalIF":3.1000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Statistical analysis of the unique characteristics of secondary structures in proteins\",\"authors\":\"Nitin Kumar Singh , Manish Agarwal , Mithun Radhakrishna\",\"doi\":\"10.1016/j.compbiolchem.2024.108237\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Protein folding is a complex process influenced by the primary sequence of amino acids. Early studies focused on understanding whether the specificity or the conservation of properties of amino acids was crucial for folding into secondary structures such as <span><math><mi>α</mi></math></span>-helices, <span><math><mi>β</mi></math></span>-sheets, turns, and coils. However, with the advent of artificial intelligence (AI) and machine learning (ML), the emphasis has shifted towards the precise nature and occurrence of specific amino acids. In our study, we analyzed a large set of proteins from diverse organisms to identify unique features of secondary structures, particularly in terms of the distribution of polar, non-polar, and charged amino acid residues. We found that <span><math><mi>α</mi></math></span>-helices tend to have a higher proportion of charged and non-polar groups compared to other secondary structures and that the presence of oppositely charged amino acid residues in helices stabilizes them, facilitating the formation of longer helices. These characteristics are distinct to <span><math><mi>α</mi></math></span>-helices. This study offers valuable insights for researchers in the field of protein design, enabling the de-novo creation of short helical peptides for a range of applications. We have also developed a web server for extensive analysis of proteins from different databases. The web server is housed at <span><span>https://proseqanalyser.iitgn.ac.in/</span><svg><path></path></svg></span></div></div>\",\"PeriodicalId\":10616,\"journal\":{\"name\":\"Computational Biology and Chemistry\",\"volume\":\"113 \",\"pages\":\"Article 108237\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational Biology and Chemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1476927124002251\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124002251","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/5 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
Statistical analysis of the unique characteristics of secondary structures in proteins
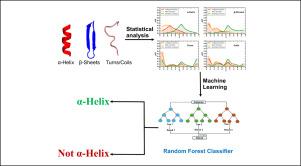
Protein folding is a complex process influenced by the primary sequence of amino acids. Early studies focused on understanding whether the specificity or the conservation of properties of amino acids was crucial for folding into secondary structures such as -helices, -sheets, turns, and coils. However, with the advent of artificial intelligence (AI) and machine learning (ML), the emphasis has shifted towards the precise nature and occurrence of specific amino acids. In our study, we analyzed a large set of proteins from diverse organisms to identify unique features of secondary structures, particularly in terms of the distribution of polar, non-polar, and charged amino acid residues. We found that -helices tend to have a higher proportion of charged and non-polar groups compared to other secondary structures and that the presence of oppositely charged amino acid residues in helices stabilizes them, facilitating the formation of longer helices. These characteristics are distinct to -helices. This study offers valuable insights for researchers in the field of protein design, enabling the de-novo creation of short helical peptides for a range of applications. We have also developed a web server for extensive analysis of proteins from different databases. The web server is housed at https://proseqanalyser.iitgn.ac.in/
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
